CD169 is one of the putative receptors of porcine reproductive and respiratory syndrome virus, also plays a major role in PRRSV infection. Computational methods including, homology modelling, molecular docking analysis and molecular dynamics simulations carried out to investigate 3D structure and potent inhibitors of CD16. Homology modelling and molecular docking were done by Maestro 10.6. A 3D structure of CD169 was obtained through homology modelling. It was later subjected protein-ligand interaction by molecular docking study. The docking results showed top ten hits compounds with the docking score energies, among those compounds MOL002433 (3R,8S,9R,10R,13R,14S,17R)-3-hydroxy-4,4,9,13,14-pentamethyl-17-[(E,2R)-6-methyl-7-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-1,2,3,7,8,10,12,15,16,17-decahydr) had the best docking score energy -8.095 kcal/mol and showed significant binding affinity and interactions with CD169 receptor active site, respectively form H bond with residues ASP-40, SER-104, LYS-107 and ASN-92. Furthermore, MD (molecular dynamics) simulations were performed by Amber 16 to investigate the stability of a ligand-protein complex. The analysis of root mean square deviation (RMSD) of CD169 /(3R,8S,9R,10R,13R,14S,17R)-3-hydroxy-4,4,9,13,14-pentamethyl-17-[(E,2R)-6-methyl-7-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-1,2,3,7,8,10,12,15,16,17-decahydr) complex revealed that CD169 protein has more stability when it interacts with the inhibitor. These findings have given us a better understanding of the functional properties and the reaction mechanism of CD169 receptor. Our results will help to identify new leads for drug discovery in PRRSV infection.
| Published in | Journal of Drug Design and Medicinal Chemistry (Volume 10, Issue 2) |
| DOI | 10.11648/j.jddmc.20241002.11 |
| Page(s) | 45-53 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2024. Published by Science Publishing Group |
PRRSV, Homology Modelling, Molecular Docking, MD Simulation, CD169
SN | Molecule Names | Structure | Docking scores/ kcal/mol | Protein-ligand interactions |
|---|---|---|---|---|
1 | (3R,8S,9R,10R,13R,14S,17R)-3-hydroxy-4,4,9,13,14-pentamethyl-17-[(E,2R)-6-methyl-7-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-1,2,3,7,8,10,12,15,16,17-decahydr |
| -8.095 | Lys107, Asn92, Asp40, Ser104 |
2 | (2R)-3-(3,4-dihydroxyphenyl)-2-[(Z)-3-(3,4-dihydroxyphenyl)acryloyl]oxy-propionic acid |
| -7.722 | Ser94, Ser20, Arg94, Ser104 |
3 | Dichotomoside |
| -7.502 | Arg94, Ser42, Ser104 |
4 | (E,E)-3,5-Di-O-caffeoylquinic acid |
| -7.407 | Val106, Arg45, Arg94, Arg104, Tyr41 |
5 | Mucic acid 1,4-lactone 5-0-gallate |
| -7.240 | Arg45, Arg94, Hie60, Glu96, Tyr38 |
6 | (-)-Maackiain-3-O-glucosyl-6'-O-malonate |
| -7.164 | Arg102, Gly100 |
7 | Luteolin-4′-glucoside |
| -7.003 | Asn92, ARG102, Ser104, Arg94 |
8 | Picraquassioside C |
| -6.845 | Ser104, Tyr41, Gly96, Arg92 |
9 | Kushenol O |
| -6.826 | Lys44, Gyl43, Tyr38, Arg52, Glu58 |
10 | Citrusin B |
| -6.725 | Tyr41, Arg94, Val106, Asn92, Arg102, Glu99 |
MD | Molecular Dynamics |
RMSD | Root Mean Square Deviation |
TCM | Traditional Chinese Medicines |
TCMSP | Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform |
PRRS | Porcine Reproductive and Respiratory Syndrome |
ORFs | Open Reading Frames |
| [1] | Jie, H., Rui, L., Hongfang, M., Songlin, Q. J. F. Structural prediction of porcine sialoadhesin V-set Ig-like domain sheds some light on its role in porcine reproductive and respiratory syndrome virus (PRRSV) infection. Front. Agr. Sci. Eng. 2016, 3(1): 65–71. |
| [2] | Holtkamp, D. J., Kliebenstein, J. B., Neumann, E., Zimmerman, J. J., Rotto, H., Yoder, T. K., et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. Journal of Swine Health and Production 2013. 21(2), 72. |
| [3] | Lunney, J. K., Fang, Y., Ladinig, A., Chen, N., Li, Y., Rowland, B., et al. Porcine reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annual Review of Animal Biosciences 2016.4, 129-154. |
| [4] | Huang, C., Zhang, Q., and Feng, W.-h. J. V. r. Regulation and evasion of antiviral immune responses by porcine reproductive and respiratory syndrome virus. Virus Vesearch 2015, 202, 101-111. |
| [5] | Du, T., Shi, Y., Xiao, S., Li, N., Zhao, Q., Zhang, A., et al. Curcumin is a promising inhibitor of genotype 2 porcine reproductive and respiratory syndrome virus infection. BMC Veterinary Research 2017, 13(1), 298. |
| [6] | Gao, J., Xiao, S., Xiao, Y., Wang, X., Zhang, C., Zhao, Q., et al. MYH9 is an essential factor for porcine reproductive and respiratory syndrome virus infection. Scientific Reports 2016, 6, 25120. |
| [7] | Fraser, I., and Gordon, S. J. E. j. o. c. b. Murine erythroleukemia (MEL) cells bear ligands for the sialoadhesin and erythroblast receptor macrophage hemagglutinins. European journal of cell biology 1994, 64(2), 217-221. |
| [8] | Guo, C., Zhu, Z., Wang, X., Chen, Y., and Liu, X. J. V. M. Pyrithione inhibits porcine reproductive and respiratory syndrome virus replication through interfering with NF-κB and heparanase. Veterinary Microbiology 2017, 231-239. |
| [9] | Crocker, P. R., Hartnell, A., Munday, J., and Nath, D. J. G. j. The potential role of sialoadhesin as a macrophage recognition molecule in health and disease. Glycoconjugate journal 1997, 601-609. |
| [10] | Delputte, P. L., and Nauwynck, H. "Porcine Arterivirus Entry in Macrophages: Heparan Sulfate–Mediated Attachment, Sialoadhesin-Mediated Internalization, and a Cell-Specific Factor Mediating Virus Disassembly and Genome Release," in The Nidoviruses. Springer) 2006, 247-252. |
| [11] | Van Breedam, W., Van Gorp, H., Zhang, J. Q., Crocker, P. R., Delputte, P. L., and Nauwynck, H. J. J. P. p. The M/GP5 glycoprotein complex of porcine reproductive and respiratory syndrome virus binds the sialoadhesin receptor in a sialic acid-dependent manner. PLoS Pathogens 2010, 6(1), e1000730. |
| [12] | Messaoudi, A., Belguith, H., Hamida, J. B. J. T. B., and Modelling, M. Homology modeling and virtual screening approaches to identify potent inhibitors of VEB-1 β-lactamase. Theoretical Biology and Medical Modelling 2013, 10(1), 22. |
| [13] | Cui, W.-Q., Qu, Q.-W., Wang, J.-P., Bai, J.-W., Bello-Onaghise, G. s., Li, Y.-A., et al. Discovery of Potential Anti-infective Therapy Targeting Glutamine Synthetase in Staphylococcus xylosus. Frontiers in Chemistry 2019, 7. |
| [14] | Shen, J., Zhang, W., Fang, H., Perkins, R., Tong, W., and Hong, H. (Year). "Homology modeling, molecular docking, and molecular dynamics simulations elucidated α-fetoprotein binding modes", in: BMC bioinformatics: BioMed Central), S6. |
| [15] | Kuntal, B. K., Aparoy, P., and Reddanna, P. J. B. r. n. EasyModeller: A graphical interface to MODELLER. BMC Research notes 2010, 3(1), 226. |
| [16] | Krieger, E., Nabuurs, S. B., and Vriend, G. Homology modeling. Methods Biochem. Anal 2003, 44, 509–523. |
| [17] | Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. J. J. o. a. c. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography 1993, 26(2), 283-291. |
| [18] | Colovos, C., and Yeates, T. O. J. P. s. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Science (1993) 2(9), 1511-1519. |
| [19] | Lüthy, R., Bowie, J. U., and Eisenberg, D. J. N. Assessment of protein models with three-dimensional profiles. Nature 1992, 356(6364), 83. |
| [20] | Lu, P., Chen, J., Zhao, H., Gao, Y., Luo, L., Zuo, X., et al. In silico syndrome prediction for coronary artery disease in traditional chinese medicine. Evidence-based Complementary and Alternative Medicine 2012. |
| [21] | Usman, M. M., Bharbhuiya, T. K., Mondal, S., Rani, S., Kyal, C., and Kumari, R. J. Combined protein and ligand based physicochemical aspects of molecular recognition for the discovery of CDK9 inhibitor. Gene Reports 2018, 13, 212-219. |
| [22] | Case, D. A., Cheatham III, T. E., Darden, T., Gohlke, H., Luo, R., Merz Jr, K. M.,... Woods. The Amber biomolecular simulation programs. J Comput Chem. 2005 December; 26(16): 1668–1688. |
| [23] | Pearlman, D. A., Case, D. A., Caldwell, J. W., Ross, W. S., Cheatham III, T. E., DeBolt, S. Kollman. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Computer Physics Communications Volume 91, Issues 1–3, 2 September 1995, Pages 1-41. |
| [24] | Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., Simmerling. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 2015, 11(8), 3696-3713. |
| [25] | Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, Da. Development and testing of a general amber force field. J Comput Chem. 2004 25(9), 1157-1174. |
| [26] | Fothergill, A. W. Antifungal susceptibility testing: clinical laboratory and standards institute (CLSI) methods. In Interactions of yeasts, moulds, and antifungal agents 2012 (pp. 65-74): Springer. |
| [27] | Xiang, Z. Advances in homology protein structure modeling. Current Protein and Peptide Science 2006, 7(3), 217-227. |
| [28] | Suri, S., Waseem, R., Bandi, S., & Shaik, S. Homology Modeling of Mus Musculus CDK5 and Molecular Docking Studies with Flavonoids. International Journal of Pharmaceutical and Clinical Research 2017; 9(6): 480-484. |
| [29] | May, A., Robinson, R., Vinson, M., Crocker, P., & Jones, E. Crystal structure of the N-terminal domain of sialoadhesin in complex with 3′ sialyllactose at 1.85 Å resolution. Molecular Cell 1998, 1(5), 719-728. |
| [30] | Attrill, H., Takazawa, H., Witt, S., Kelm, S., Isecke, R., Brossmer, R., Crocker. The structure of siglec-7 in complex with sialosides: leads for rational structure-based inhibitor design. Biochem. J. 2006, 397(2), 271-278. |
| [31] | Itteboina, R., Ballu, S., Sivan, S. K., Manga, V. J. Molecular docking, 3D-QSAR, molecular dynamics, synthesis and anticancer activity of tyrosine kinase 2 (TYK 2) inhibitors. Journal of Receptors and Signal Transduction 2018, 38(5-6), 462-474. |
APA Style
Eliphaz, N., Cui, W., Xiao, H., Bello-Onaghise, G., Yang, T., et al. (2024). Homology Modelling and Molecular Docking Studies to Discover Potent Inhibitors of CD169 in PRRSV Infection. Journal of Drug Design and Medicinal Chemistry, 10(2), 45-53. https://doi.org/10.11648/j.jddmc.20241002.11
ACS Style
Eliphaz, N.; Cui, W.; Xiao, H.; Bello-Onaghise, G.; Yang, T., et al. Homology Modelling and Molecular Docking Studies to Discover Potent Inhibitors of CD169 in PRRSV Infection. J. Drug Des. Med. Chem. 2024, 10(2), 45-53. doi: 10.11648/j.jddmc.20241002.11
AMA Style
Eliphaz N, Cui W, Xiao H, Bello-Onaghise G, Yang T, et al. Homology Modelling and Molecular Docking Studies to Discover Potent Inhibitors of CD169 in PRRSV Infection. J Drug Des Med Chem. 2024;10(2):45-53. doi: 10.11648/j.jddmc.20241002.11
@article{10.11648/j.jddmc.20241002.11,
author = {Nsabimana Eliphaz and Wen-qiang Cui and Han Xiao and God’spower Bello-Onaghise and Tang Yang and Yu Fei and Zhang Yue Feng and Jun-jie Qin and Wen-xin Guo and Yan-hua Li},
title = {Homology Modelling and Molecular Docking Studies to Discover Potent Inhibitors of CD169 in PRRSV Infection
},
journal = {Journal of Drug Design and Medicinal Chemistry},
volume = {10},
number = {2},
pages = {45-53},
doi = {10.11648/j.jddmc.20241002.11},
url = {https://doi.org/10.11648/j.jddmc.20241002.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.jddmc.20241002.11},
abstract = {CD169 is one of the putative receptors of porcine reproductive and respiratory syndrome virus, also plays a major role in PRRSV infection. Computational methods including, homology modelling, molecular docking analysis and molecular dynamics simulations carried out to investigate 3D structure and potent inhibitors of CD16. Homology modelling and molecular docking were done by Maestro 10.6. A 3D structure of CD169 was obtained through homology modelling. It was later subjected protein-ligand interaction by molecular docking study. The docking results showed top ten hits compounds with the docking score energies, among those compounds MOL002433 (3R,8S,9R,10R,13R,14S,17R)-3-hydroxy-4,4,9,13,14-pentamethyl-17-[(E,2R)-6-methyl-7-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-1,2,3,7,8,10,12,15,16,17-decahydr) had the best docking score energy -8.095 kcal/mol and showed significant binding affinity and interactions with CD169 receptor active site, respectively form H bond with residues ASP-40, SER-104, LYS-107 and ASN-92. Furthermore, MD (molecular dynamics) simulations were performed by Amber 16 to investigate the stability of a ligand-protein complex. The analysis of root mean square deviation (RMSD) of CD169 /(3R,8S,9R,10R,13R,14S,17R)-3-hydroxy-4,4,9,13,14-pentamethyl-17-[(E,2R)-6-methyl-7-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-1,2,3,7,8,10,12,15,16,17-decahydr) complex revealed that CD169 protein has more stability when it interacts with the inhibitor. These findings have given us a better understanding of the functional properties and the reaction mechanism of CD169 receptor. Our results will help to identify new leads for drug discovery in PRRSV infection.
},
year = {2024}
}
TY - JOUR T1 - Homology Modelling and Molecular Docking Studies to Discover Potent Inhibitors of CD169 in PRRSV Infection AU - Nsabimana Eliphaz AU - Wen-qiang Cui AU - Han Xiao AU - God’spower Bello-Onaghise AU - Tang Yang AU - Yu Fei AU - Zhang Yue Feng AU - Jun-jie Qin AU - Wen-xin Guo AU - Yan-hua Li Y1 - 2024/08/06 PY - 2024 N1 - https://doi.org/10.11648/j.jddmc.20241002.11 DO - 10.11648/j.jddmc.20241002.11 T2 - Journal of Drug Design and Medicinal Chemistry JF - Journal of Drug Design and Medicinal Chemistry JO - Journal of Drug Design and Medicinal Chemistry SP - 45 EP - 53 PB - Science Publishing Group SN - 2472-3576 UR - https://doi.org/10.11648/j.jddmc.20241002.11 AB - CD169 is one of the putative receptors of porcine reproductive and respiratory syndrome virus, also plays a major role in PRRSV infection. Computational methods including, homology modelling, molecular docking analysis and molecular dynamics simulations carried out to investigate 3D structure and potent inhibitors of CD16. Homology modelling and molecular docking were done by Maestro 10.6. A 3D structure of CD169 was obtained through homology modelling. It was later subjected protein-ligand interaction by molecular docking study. The docking results showed top ten hits compounds with the docking score energies, among those compounds MOL002433 (3R,8S,9R,10R,13R,14S,17R)-3-hydroxy-4,4,9,13,14-pentamethyl-17-[(E,2R)-6-methyl-7-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-1,2,3,7,8,10,12,15,16,17-decahydr) had the best docking score energy -8.095 kcal/mol and showed significant binding affinity and interactions with CD169 receptor active site, respectively form H bond with residues ASP-40, SER-104, LYS-107 and ASN-92. Furthermore, MD (molecular dynamics) simulations were performed by Amber 16 to investigate the stability of a ligand-protein complex. The analysis of root mean square deviation (RMSD) of CD169 /(3R,8S,9R,10R,13R,14S,17R)-3-hydroxy-4,4,9,13,14-pentamethyl-17-[(E,2R)-6-methyl-7-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-1,2,3,7,8,10,12,15,16,17-decahydr) complex revealed that CD169 protein has more stability when it interacts with the inhibitor. These findings have given us a better understanding of the functional properties and the reaction mechanism of CD169 receptor. Our results will help to identify new leads for drug discovery in PRRSV infection. VL - 10 IS - 2 ER -
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
Veterinary Medicine Engineering Laboratory, Beijing Centre Technology Co., Ltd. Beijing, China
Heilongjiang Provincial Agricultural Products and Veterinary Medicine Technical Appraisal Station, Harbin, China
College of Veterinary Medicine, Northeast Agricultural University, Harbin, China; Heilongjiang Key Laboratory for Animal Disease Control and Pharmaceutical Development, Harbin, China
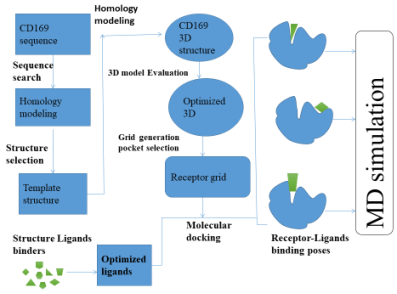
Figure 1. The work flow applied to discover the novel CD169 inhibitor.
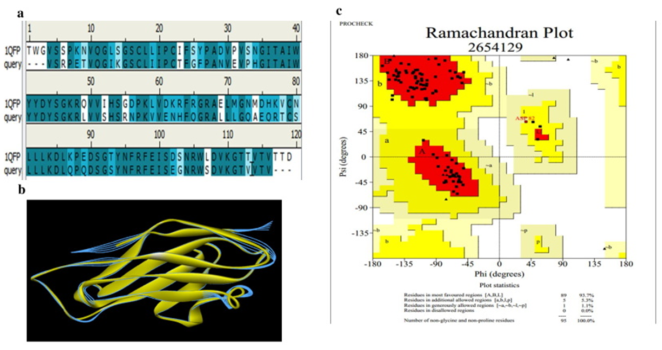
Figure 2. (a) Comparison of sequence alignment between model (CD19) and template (1QFP). (b) Superposition of the target (CD169) and template (1QFP) (RMSD: 0.359 Å). (c) Ramachandran plot ‘calculation of CD169 model provided by PROCHECK: 93% residues in favorable regions; 5.3% residues in additional allowed regions; 1.1% residues in generously allowed regions; 0% residues in disallowed regions. Regions; 1.1% residues in generously allowed regions; 0% residues in disallowed regions.
Figure 3. (a) Protein-ligand interactions (binding mode) of MOL002433 against CD169. MOL002433 was shown in a aqua color stick model. The binding site residues are shown as a stick model lavender color. The yellow dots indicate H bonds. (b) The protein-ligand interactions are shown in surface. MOL002433 was shown in aqua color stick mode, and CD169 was shown in an electrostatic surface.
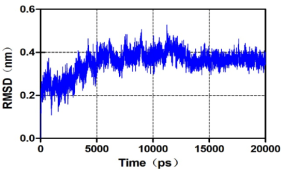
Figure 4. The molecular dynamics (MD) simulation of the CD169/MOL002433 complex. Shows the root-mean-square deviation (RMSD) calculated for the backbone atoms of the protein during MD simulation.
Information











