Coumarins of natural origin have been explored as potential inhibitors of P-glycoprotein (P-gp). Esculetin which belongs to the class of coumarin has been derivatized with known hydrazine pharmacophores viz; benzoyl hydrazine (BH), isonicotinyl hydrazine (INH), and hydrazino benzoic acid. The homology modeling approach was used to predict the three-dimensional structure of human P-gp. An in-silico study has been performed for the structural insight into the molecular mechanism of P-gp inhibition of the esculetin derivatives by molecular docking (MD) and simulation studies. The cell cytotoxic activities of the synthesized compounds were evaluated using in-vitro studies. The sublines resistant doxorubicin (MCF-7/R) were generated and the activities of P-gp proteins were estimated using fluorescent dye accumulation assays. The E-BH showed promising P-gp inhibitory activity and cell cytotoxicity against MCF7 and MCF7/R (resistant) breast cancer cell lines. In line with experimental observations, the E-BH (Esculetin benzoyl hydrazine) has yielded the lowest energy stable complex with P-gp and is stabilized by intermolecular hydrogen bonding and more hydrophobic interactions during 100 ns of simulation. This suggested that the activity of P-gp is probably controlled by hydrophobic interactions. Performed experimental and computational studies has helped to elucidate the mechanism of P-gp inhibition by E-BH. Thus, amongst the three derivatives; E-BH exhibits greater efficacy in blocking the efflux mechanism.
| Published in | American Journal of Biomedical and Life Sciences (Volume 12, Issue 3) |
| DOI | 10.11648/j.ajbls.20241203.12 |
| Page(s) | 30-48 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2024. Published by Science Publishing Group |
Esculetin Hydrazine Derivatives, Pgp Inhibitors, Homology Modeling, Docking, Molecular Dynamics Simulation
Drug | M of Esculetin derivatives (IC50) |
|---|---|
Esculetin | 135.5±22.6 |
E-INH | 16.3±1.6 |
E-BH | 8.4±1.83 |
E-HBA | 11.6±2.1 |
Verapamil | 15.1±2.3 |
Amino acid regions | Transmembrane Domains No. | Amino acid regions | Loop/Turns |
|---|---|---|---|
1-86 | Helices/TM1 | 87-96 | Loop |
97-164 | TM2 | 165-169 | Loop |
170-210 | TM3 | 211-212 | Loop |
213-267 | TM4 | 268-269 | Loop |
270-322 | TM5 | 323-329 | Loop |
330-369 | TM6 | 381-626 | NBD1/Helices/Sheets/Loops |
627-683 | Linker Region | 684-698 | Loop |
699-740 | TM7 | 741-746 | Loop |
747-797 | TM8 | 798-810 | Loop |
811-852 | TM9 | 853-855 | Loop |
856-909 | TM10 | 910-912 | Loop |
913-966 | TM11 | 967-976 | Loop |
971-1013 | TM12 | 1014-1026 | Loops |
1027-1280 | NBD2 |
Sr. No. | Atoms Involved 1-2-3 in hydrogen bonding | Distance in Å | Binding Energy in kcal/mol | Fig. Ref. |
|---|---|---|---|---|
Benzoyl Hydrazide (E-BH) with P-glycoprotein | ||||
1 | Tyr307-OH.....O4-E-BH | 2.53 | -9.02 | 7B |
2 | Gln725-NE2......O4-E-BH | 2.72 | ||
3 | Phe728, Ala729, Phe732, Leu975, Phe978, Val982 | Hydrophobic interactions | ||
4-Hydrazino Benzoic Acid (E-HBA) with P-glycoprotein | ||||
4 | Tyr307-OH.....O3-E-HBA | 2.76 | -8.60 | S8-A |
5 | Gln725-NE2......O2-E-HBA | 2.59 | ||
6 | Phe72, Phe728, Ala729, Phe732, Leu975, Phe978, Ser979, Val982 | Hydrophobic interactions | ||
Isonicotyl Hydrazide (E-INH) with P-glycoprotein | ||||
7 | Gln725-NE2......O3-E-INH | 2.67 | -8.11 | S8-B |
8 | Gln725-NE2......O4-E-INH | 2.44 | ||
9 | Phe72, Phe728, Ala729, Phe732, Ser733, Ile736, Leu975, Phe978, Val982 | Hydrophobic interactions | ||
Esculetin (E) with P-glycoprotein | ||||
10 | Tyr307-OH.....O3-Esculetin | 2.83 | -6.27 | S8-C |
11 | Tyr307-OH.....O4-Esculetin | 2.61 | ||
12 | Tyr310-OH.......O2-Esculetin | 3.15 | ||
Gln725-NE2......O3-Esculetin | 2.57 | |||
Phe336, Phe728 | Hydrophobic interactions | |||
E | Esculetin |
E-BH | Esculetin Benzoyl Hydrazine |
E-INH | Esculetin Isonicotinyl Hydrazine |
E-HBA | Esculeting Hydrazine Benzoic Acid |
P-gp | P-glycoprotein |
MCF-7/R | MCF-7/Resistant Cell Lines Expressing P-gp |
MDR | Multidrug Resistance |
MDCK | Madin Darby Canine Kidney Cells |
FTIR | Fourier Transform Infrared Spectroscopy |
NMR | Nuclear Magnetic Resonance |
ANOVA | Analysis of Variance |
NCCS | National Centre for Cell Sciences |
DFT | Density Functional Theory |
LINCS | Linear Constraint Solver for Molecular Simulations |
VMD | Visual Molecular Dynamics |
MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
DOPE | Discrete Optimized Protein Energy |
| [1] | M. M. Gottesman, C. A. Hrycyna, P. V. Schoenlein, U. A. Germann, I Pastan, Genetic analysis of the multidrug transporter, Annu. Rev. Genet. 29 (1995) 607-649. |
| [2] | T. W. Loo, D. M. Clarke, Identification of residues in the drug-binding site of human P- glycoprotein using a thiol-reactive substrate, J. Biol. Chem. 272 (1997) 31945-31948. |
| [3] | S. V. Ambudkar, G Kimchi-Sarfaty, Z. E. Suana, M. M. Gottesman, P-glycoprotein: from genomics to mechanism, Oncogene 22 (2003) 7468-7485. |
| [4] | P. D. Eckford, F. J. Sharom, ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109 (2009) 2989-3011. |
| [5] | B Marquez, B. F. Van, ABC multidrug transporter: Target for modulation of drug pharmacokinetics and drug-drug interactions. Curr. drug targets 12 (2011) 600-620. |
| [6] | S Kunjachan, B Rychlik, G Storm, F Kiessling, T. Lammers, Multidrug resistance: Physiological principles and nanomedical solutions, Adv. Drug Deliv. Rev. 65 (2013) 1852-1865. |
| [7] | M. Leopoldo, P. Nardulli, M. Contino, F. Leonetti, G. Luurtsema, N. A. Colabufo. An updated patent revire on P-glycoprotein inhibitors, Expert Opin. Ther. Pat. 29 (2019) 455-461. |
| [8] | J. Marcoux, S. C. Wang, A. Ploitis, et al., Mass spectrometry reveals synergetic effects to nucleotides, lipids, and drug binding to a multidrug resistance efflux pump, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 9704-9709. |
| [9] | D. Waghray, Q. Zhang, Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment, J. Med. Chem. 61(2018) 5108-5121. |
| [10] | E. N. Yakusheva, D. S. Titov, Structure and Function of Multidrug Resistance Protein-1 Biochemistry (Mosc) 83(2018) 907-929. |
| [11] | S. Mollazadeh, A. Sahebkar, F. Hadizadeh, J. Behravan, S. Arabzadeh, Structural and functional aspects of P-glycoprotein and its inhibitors, Life Sci. 214 (2018) 118-123. |
| [12] | K. P. Sigdel, L. A. Wilt, B. P. Marsh, A. G. Roberts, G. M. King, The conformation and dynamics of P-glycoprotein in a lipid bilayer investigated by atomic force microscopy, Biochem. Pharmacol. 156 (2018) 302-311. |
| [13] | Sajid, N. Raju, S. Lusvarghi, S. Vahedi, R. E. Swenson, S. V. Ambudkar, Synthesis and characterization of Bodpy-FL-cyclosporin A as a substrate for multidrug resistance-linked P-glycoprotein (ABCB1), Drug Metab. Dispos. 47(2019) 1013-1023. |
| [14] | W. Guo, W. Dong, M. Li, Y. Shen, Mitochondria P-glycoprotein confers paclitaxel resistance on ovarian cancer cells, Onco. Targets Ther. 12 (2019) 3881-3891. |
| [15] | F. Montanari, G. F. Ecker, Prediction of drug-ABC-transporter interaction - Recent advances and future challenges, Adv. Drug Deliv. Rev. 86 (2015) 17-26. |
| [16] | J. W. McCormick, P. D. Vogel, J. G. Wise, Multiple drug transport through Human P-Glycoprotein, Biochemistry 54 (2015) 4374-4390. |
| [17] | J. Bender, J. Fang, R. Simon, A computational study of the inhibition mechanisms of P-glycoprotein mediated paclitaxel efflux by kinase inhibitors, BMC Syst. Biol. 11 (2017) 1-10. |
| [18] | J. Wang, N. Seebacher, H. Shi, Q. Kan, Z. Duan, Novel strategies to prevent the development of multidrug resistance (MDR) in cancer, Oncotarget 8 (2017) 84559-84571. |
| [19] | S. Varghese, A. Palaniappan, Computational Pharmacogenetics of P-Glycoprotein Mediated Antiepileptic Drug Resistance. Open Bioinforma J. 11 (2018) 197- 207. |
| [20] | L. Domicevica, H. Koldso, P. C. Biggin, Multiscale molecular dynamics simulations of lipid interactions with P-glycoprotein in a complex membrane, J. Mol. Graph. Model. 80 (2018) 147-156. |
| [21] | E. Barreto-Ojeda, V. Corradi, R-X. Gu, D. P. Tieleman, Coarse-grained molecular dynamics simulations reveal lipid access pathways in P-glycoprotein, J. Gen. Physiol. 150 (2018) 417-429. |
| [22] | L. Wang, L. Zhang, F. Liu, Y. Sun, Molecular energetics of Doxorubicin pumping by human P-glycoprotein, J. Chem. Inf. Model. 59 (2019) 3889-3898. |
| [23] | Z. Bikadi, I. Hazal, D. Malik, K. Jemnitz, Z. Veres, P. Hari, Z. Ni, T. W. Loo, D. M. Clarke, E. Hazai, Q. Mao, Predicting P-glycoprotein-mediated drug transport based on support vector macnine and three dimensional crystal structure of P-glycoprotein, Plos One 6 (2011) e25815. |
| [24] | L. Chen, Y. Li, H. Yu, L. Zhang, T. Hou, Computational models for predicting substrates or inhibitors of P-glycoprotein, Drug Discov. Today 17(2012) 343-51. |
| [25] | R. Prajapati, A. T. Sangamwar, Translocation mechanism of P-glycoprotein and conformational changes occurring at drug-binding site: Insights from multi-targeted molecular dynamics, Biochim. Biophys. Acta Biomembr. 1838 (2014) 2882-2898. |
| [26] | M. L. Gonzalez, D. M. A. Vera, J. Laiolo, M. B. Joray, M. Maccioni, S. M. Palacios, G. M. Molina, P. A. Lanza, S. Gancedo, V. Rumjanek, M. C. Carpinella, Mechanism underlying the reversal of drug resistance in P-Glycoprotein-expressing leukemia cells by pinoresinol and the study of a derivative, Front. Pharmacol. 25 (2017) 205. |
| [27] | Cui, C. Y. Cai, H. L. Gao, L. Ren, N. Ji, P. Gupta, Y. Yang, S. Shukla, S. V. Ambudkar, D. H. Yang, Z. S. Chen Glesatinib, a c-MET/SMO Dual Inhibitor, Antagonizes P-glycoprotein mediated multidrug resistance in cancer cells, Front. Oncol. 25 (2019) 313. |
| [28] | J. G. Wise, Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites, Biochemistry 51 (2012) 5125-5141. |
| [29] | P. C. Wen, B. Verhalen, S. Wilkens, H. S. Nchaourab, E. Tajkhorshid, On the origine large flexibility of P-glycoprotein in the inward facing state, J. Biol. Chem. 288 (2013) 19211-19220. |
| [30] | J. P. Becker, G. Depret, F. VanBambeke, P. M. Tulkens, M. Prevost, Molecular models of human P-glycoprotein in two different catalytic states, BMC Struct. Biol. 9 (2009) 3.31. J. R. |
| [31] | Daddam, M. R. Dowlathabad, S. Panthangi, P. Jasti, Molecular docking and P-glycoprotein inhibitory activity of flavonoids, Interdiscip Sci. 6(2014) 167-175. L. |
| [32] | Domicevica, P. C. Biggin, Homology modelling of human P-glycoprotein, Biochem. Soc. Trans. 43 (2015) 952-958. |
| [33] | A. Tripathi, K. Misra, Inhibition of P-Glycoprotein Mediated Efflux of Paclitaxel by Coumarin Derivatives in Cancer Stem Cells: An In Silico Approach, Comb Chem High Throughput Screen. 19(2016) 497-506. |
| [34] | K. Wang, Y. Zhang, S. I. N. Ekunwe, et al., Antioxidant activity and inhibition effect on the growth of human colon carcinoma (HT-29) cells of esculetin from Cortex Fraxini, Med Chem Res. 20 (2011) 968–974. |
| [35] | Y. J. Jeon, J. Y. Jang, J. H. Shim, P. K. Myung, J. I. Chae, Esculetin, a Coumarin Derivative, Exhibits Anti-proliferative and Pro-apoptotic Activity in G361 Human Malignant Melanoma, J Cancer Prev. 20 (2015) 106-12. |
| [36] | C. Barthomeuf, J. Grassi, M. Demeule, C. Fournier, D. Boivin, R. Béliveau, Inhibition of P-glycoprotein transport function and reversion of MDR1 multidrug resistance by cnidiadin, Cancer Chemother. Pharmacol. 56 (2005) 173-81. |
| [37] | V. Kumar, Hydrazone: A promising pharmacophore in medicinal chemistry, J. Pharmacogn Phytochem. 7(2018) 40-43. |
| [38] | N. Duangdee, W. Mahavorasirikul, S. Prateeptongkum, Design synthesis and anti-proliferative activity of some new coumarin substituted hydrazide-hydrazone derivatives, J Chem Sci. 132 (2020) 66. |
| [39] | H. Morjani, N. Aouali, R. Belhoussine, R. J. Veldman, T. Levade, M. Manfait, Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets, Int. J. Cancer 94 (2001) 157-165. |
| [40] | G. Batist, A. Tulpule, B. K. Sinha, A. G. Katki, C. E. Myers, K. H. Cowan Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells, J. Biol. Chem. 261 (1986) 15544-15549. |
| [41] | T. L. Riss, R. A. Moravec, A. L. Niles, et al., Cell Viability Assays. In: Sittampalam GS, Grossman A, Brimacombe K, et al., editors. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2016. |
| [42] | V. M. Le, E. Jouan, B. Stieger, V. Lecureur, O. Fardel, Regulation of human hepatic drug transporter activity and expression by diesel exhaust particle extract, Plos One 10 (2015) e0121232. |
| [43] | B. Webb, A. Sali, Comparative Protein Structure Modeling Using MODELLER, Curr. Protoc. Bioinformatics 54 (2016) 5.6.1-5.6.37. |
| [44] | R. A. Laskowski, M. W. MacArthur, D. S. Moss, J. M. Thornton, PROCHECK a program to check the stereochemical quality of protein structures, J. Appl. Crystallography 26 (1993) 283-291. |
| [45] | M. Wiederstein, M. J. Sippl, ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins, Nucleic Acids Res. 35 (2007) W407-W410. |
| [46] | N. M. Kumbhar, S. K. Nimal, S. Barale, S, Ka.mble, R. S. Bavi, K. D. Sonawane, R. N. Gacche. Identification of novel leads as potent inhibitors of HDAC3 using ligand-based pharmacophore modeling and MD simulation, Scientific Reports, 2022, 12(1): 1712. |
| [47] | K. S. Gavale, S. R. Chavan, N. M. Kumbhar, S. Kawade, P. Doshi, A. Khan, D. D. Dhavale, α-Geminal disubstituted pyrrolidine iminosugars and their C-4-fluoro analogues: Synthesis, glycosidase inhibition and molecular docking studies, Bioorg. Med. Chem. 25 (2017) 5148-5159. |
| [48] | G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell, A. J. Olson, Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility, J. Comput. Chem. 16 (2009) 2785-2791. |
| [49] | A. D. Becke, Density functional thermochemistry. III. The role of exact exchange, J. Chem. Phys. 98 (1992). 5648-5652. |
| [50] | M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian, Inc., Wallingford CT, 2009. |
| [51] | A. C. Wallace, R. A. Laskowski, Thornton JM LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions, Protein Eng. 8 (1996) 127-134. |
| [52] | S. Jo, T. Kim, V. G. Iyer, W. Im, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem. 29 (2008) 1859-65. |
| [53] | J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks, A. D. MacKerell, J. B. Klauda, W. Im, CHARMMGUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/ OpenMM Simulations Using the CHARMM36 Additive Force Field, J. Chem. Theory Comput. 12 (2016) 405-13. |
| [54] | Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL, OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012; 40: D370-D376. |
| [55] | M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. C. Smith, B. Hess, E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX 1 (2015) 19-25. |
| [56] | B. Hess. P-LINCS: a parallel linear constraint solver for molecular simulation, J. Chem. Theor. Comput. 4 (2008) 116-122. |
| [57] | U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee, L. G. Pedersen, A smooth particle mesh Ewald method, J. Chem. Phys. 103 (1995) 8577-8593. |
| [58] | W. Humphrey, A. Dalke, K. Schulten, VMD: visual molecular dynamics, J. Mol. Graph. Model. 14 (1996) 33-38. |
| [59] | E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng, T. E. Ferrin, UCSF chimera -A visualization system for exploratory research and analysis, J. Comput. Chem. 25 (2004) 1605-1612. |
| [60] | B. G. Durie, W. S. Dalton, Reversal of drug-resistance in multiple myeloma with verapamil, Br. J. Haematol. 68 (1988) 203-206. |
| [61] | T. W. Sweatman, R. Seshadri, M. Israel, Metabolism and elimination of rhodamine 123 in the rat, Cancer Chemother. Pharmacol. 27 (1990) 205-210. |
| [62] | R. Yumoto, T. Murakami, M. Sanemasa, R. Nasu, J. Nagai, M. Takano, Pharmacokinetic interaction of cytochrome P450 3A-related compounds with rhodamine 123, a P-glycoprotein substrate, in rats pretreated with dexamethasone, Drug Metab. Dispos. 29 (2001) 145-151. PMID: 11159804 |
| [63] | Elodie Jouan 1, Marc Le Vée 1, Abdullah Mayati 1, Claire Denizot 2, Yannick Parmentier 2 and Olivier Fardel, Evaluation of P-Glycoprotein Inhibitory Potential Using a Rhodamine 123 Accumulation Assay, Pharmaceutics 2016, 8, 12; |
| [64] | A. B. Ward, P. Szewczyk, V. Grimard, Structure of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 13386-13391. |
| [65] | P. H. Palestro, L. Gavernet, G. L. Estiu, L. E. B. Bruno, Docking Applied to the Prediction of the Affinity of Compounds to P-Glycoprotein, Biomed. Res. Int. 358425 (2014) 1-10. |
| [66] | T. W. Loo, D. M. Clarke, Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane, J. Biol. Chem. 275 (2000) 39272-39278. |
| [67] | Mora Lagares L, Minovski N, Caballero Alfonso AY, et al. Homology Modeling of the Human P-glycoprotein (ABCB1) and Insights into Ligand Binding through Molecular Docking Studies. Int J Mol Sci. 2020; 21(11): 4058. Published 2020 Jun 5. |
| [68] | T. W. Loo, M. C. Bartlett, D. M. Clarke, Identification of residues in the drug translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis, J. Biol. Chem. 284 (2009) 24074-24087. |
| [69] | E. Dolghih, C. Bryant, A. R. Renslo, M. P. Jacobson, Predicting binding to P-Glycoprotein by flexible receptor docking, Plos Comput. Biol. 7 (2011) e1002083. |
| [70] | S. Kanaoka, Y. Kimura, M. Fujikawa, Y. Nakagawa, K. Ueda, M. Akamats, Substrate recognition by P-glycoprotein efflux transporters: Structure-ATPase activity relationship of diverse chemicals and agrochemicals, J. Pestic. Sci. 38(2013) 112-122. |
| [71] | W. Liu, Q. Meng, Y. Sun, C. Wang, X. Huo, Z. Liu, P. Sun, H. Sun, X. Ma, K. Liu, Targeting P-Glycoprotein: Nelfinavir Reverses Adriamycin Resistance in K562/ADR Cells, Cell Physiol. Biochem. 51 (2018) 1616-1631. |
| [72] | L. Pan, S. G. Aller, Allosteric role of substrate occupancy toward the alignment of P-glycoprotein nucleotide binding domains, Sci. Rep. 8 (2018) 14643. |
| [73] | S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, R. Zhuo, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch, G. Chang, Structure of P-glycoprotein reveals a molecular basis for poly specific drug binding, Science 323 (2009) 1718-1722. |
| [74] | Y. Raviv, H. B. Pollard, E. P. Bruggemann, I. Pastan, M. M. Gottesman, Photosensitized labeling of a functional multidrug transporter in living drug-resistant tumor cells, J. Biol. Chem. 265 (1990) 3975-80. |
APA Style
Kumbhar, N., Khan, N., Bavi, R., Barage, S., Khan, A. (2024). Reversal of P-glycoprotein Mediated Multidrug Resistance in MCF-7/R Cancer Cells by Esculetin Derivatives: Experimental and MD Simulation Studies. American Journal of Biomedical and Life Sciences, 12(3), 30-48. https://doi.org/10.11648/j.ajbls.20241203.12
ACS Style
Kumbhar, N.; Khan, N.; Bavi, R.; Barage, S.; Khan, A. Reversal of P-glycoprotein Mediated Multidrug Resistance in MCF-7/R Cancer Cells by Esculetin Derivatives: Experimental and MD Simulation Studies. Am. J. Biomed. Life Sci. 2024, 12(3), 30-48. doi: 10.11648/j.ajbls.20241203.12
AMA Style
Kumbhar N, Khan N, Bavi R, Barage S, Khan A. Reversal of P-glycoprotein Mediated Multidrug Resistance in MCF-7/R Cancer Cells by Esculetin Derivatives: Experimental and MD Simulation Studies. Am J Biomed Life Sci. 2024;12(3):30-48. doi: 10.11648/j.ajbls.20241203.12
@article{10.11648/j.ajbls.20241203.12,
author = {Navanath Kumbhar and Neelofar Khan and Rohit Bavi and Sagar Barage and Ayesha Khan},
title = {Reversal of P-glycoprotein Mediated Multidrug Resistance in MCF-7/R Cancer Cells by Esculetin Derivatives: Experimental and MD Simulation Studies
},
journal = {American Journal of Biomedical and Life Sciences},
volume = {12},
number = {3},
pages = {30-48},
doi = {10.11648/j.ajbls.20241203.12},
url = {https://doi.org/10.11648/j.ajbls.20241203.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajbls.20241203.12},
abstract = {Coumarins of natural origin have been explored as potential inhibitors of P-glycoprotein (P-gp). Esculetin which belongs to the class of coumarin has been derivatized with known hydrazine pharmacophores viz; benzoyl hydrazine (BH), isonicotinyl hydrazine (INH), and hydrazino benzoic acid. The homology modeling approach was used to predict the three-dimensional structure of human P-gp. An in-silico study has been performed for the structural insight into the molecular mechanism of P-gp inhibition of the esculetin derivatives by molecular docking (MD) and simulation studies. The cell cytotoxic activities of the synthesized compounds were evaluated using in-vitro studies. The sublines resistant doxorubicin (MCF-7/R) were generated and the activities of P-gp proteins were estimated using fluorescent dye accumulation assays. The E-BH showed promising P-gp inhibitory activity and cell cytotoxicity against MCF7 and MCF7/R (resistant) breast cancer cell lines. In line with experimental observations, the E-BH (Esculetin benzoyl hydrazine) has yielded the lowest energy stable complex with P-gp and is stabilized by intermolecular hydrogen bonding and more hydrophobic interactions during 100 ns of simulation. This suggested that the activity of P-gp is probably controlled by hydrophobic interactions. Performed experimental and computational studies has helped to elucidate the mechanism of P-gp inhibition by E-BH. Thus, amongst the three derivatives; E-BH exhibits greater efficacy in blocking the efflux mechanism.
},
year = {2024}
}
TY - JOUR T1 - Reversal of P-glycoprotein Mediated Multidrug Resistance in MCF-7/R Cancer Cells by Esculetin Derivatives: Experimental and MD Simulation Studies AU - Navanath Kumbhar AU - Neelofar Khan AU - Rohit Bavi AU - Sagar Barage AU - Ayesha Khan Y1 - 2024/08/27 PY - 2024 N1 - https://doi.org/10.11648/j.ajbls.20241203.12 DO - 10.11648/j.ajbls.20241203.12 T2 - American Journal of Biomedical and Life Sciences JF - American Journal of Biomedical and Life Sciences JO - American Journal of Biomedical and Life Sciences SP - 30 EP - 48 PB - Science Publishing Group SN - 2330-880X UR - https://doi.org/10.11648/j.ajbls.20241203.12 AB - Coumarins of natural origin have been explored as potential inhibitors of P-glycoprotein (P-gp). Esculetin which belongs to the class of coumarin has been derivatized with known hydrazine pharmacophores viz; benzoyl hydrazine (BH), isonicotinyl hydrazine (INH), and hydrazino benzoic acid. The homology modeling approach was used to predict the three-dimensional structure of human P-gp. An in-silico study has been performed for the structural insight into the molecular mechanism of P-gp inhibition of the esculetin derivatives by molecular docking (MD) and simulation studies. The cell cytotoxic activities of the synthesized compounds were evaluated using in-vitro studies. The sublines resistant doxorubicin (MCF-7/R) were generated and the activities of P-gp proteins were estimated using fluorescent dye accumulation assays. The E-BH showed promising P-gp inhibitory activity and cell cytotoxicity against MCF7 and MCF7/R (resistant) breast cancer cell lines. In line with experimental observations, the E-BH (Esculetin benzoyl hydrazine) has yielded the lowest energy stable complex with P-gp and is stabilized by intermolecular hydrogen bonding and more hydrophobic interactions during 100 ns of simulation. This suggested that the activity of P-gp is probably controlled by hydrophobic interactions. Performed experimental and computational studies has helped to elucidate the mechanism of P-gp inhibition by E-BH. Thus, amongst the three derivatives; E-BH exhibits greater efficacy in blocking the efflux mechanism. VL - 12 IS - 3 ER -
Department of Biotechnology, Medical Information Management, Shivaji University, Kolhapur, India
Biography: Navanath Kumbhar is a Research Scholar at Department of Biotechnology, Medical Information Management, Shivaji University, Kolhapur, India. He has completed his doctoral work in Bioinformatics and Computational Biology in 2012, from Shivaji University Kolhapur. His expertise are conformational energy calculations of tRNA modifications using Semi-empircal PM6 and geometry optimizations using DFT, HF and MO6 methods were performed.. Homolegy modeling, molecular docking and MD simulations of P-glycoprotein (MDR), Synthetic iminosugars have been performed. The structural details of Fluorinated sugar amino acids peptides were predicted using DFT studies. subunit.

Department of Chemistry, Savitribai Phule Pune University, Pune, India
Department of Biomedical Engineering, China Pharmaceutical University, Nanjing, China; School of Chemical Science, Punyashlok Ahilyadevi Holkar Solapur University, Solapur, India
Biography: Rohit Bavi is a Postdoctoral Research Associate at China Pharmaceutical University. He has completed his doctoral studies from Shivaji University, Kolhapur in 2014. He is an expert in Schrodinger, Autodoc and structural biology.

Amity Institute of Biotechnology, Amity University, Mumbai, India
Biography: Sagar Barage is an Associate Professor in Amity Institute of Biotechnology, Mumbai. He has completed his doctoral studies from Shivaji University, Kolhapur in 2014. His current research interest include Bioinformatics, Computational Biology, Drug designing, Molecular Modelling.

Department of Chemistry, Savitribai Phule Pune University, Pune, India
Biography: Ayesha Khan (nee Sabari Dutta) is an Assistant Professor in Savitribai Phule Pune University Department of Chemistry. She has completed her doctoral studies from Pune University. She has completed her postodoctoral research from School of medicine, Wayne State University, Michigan, USA. Her current research interest include Drug designing, Microbiology and animal tissue culture. Ms. Neelofar Khan was a Project Assistant at Institute of Bioinformatics and Biotechnology who synthesized the esculetin compounds.

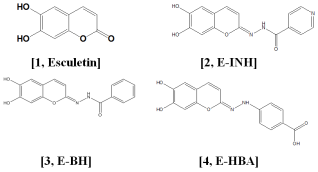
Scheme 1. Esculetin derivatives.
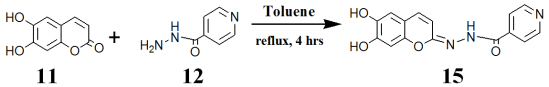
Scheme 2. Preparation of esculetin-isonicotyl hydrazide.
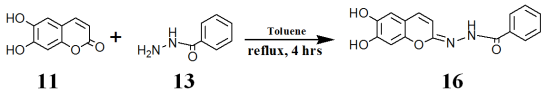
Scheme 3. Preparation of esculetin-benzoyl hydrazide.
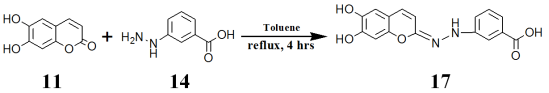
Scheme 4. Preparation of esculetin- hydrazinobenzoic acid.
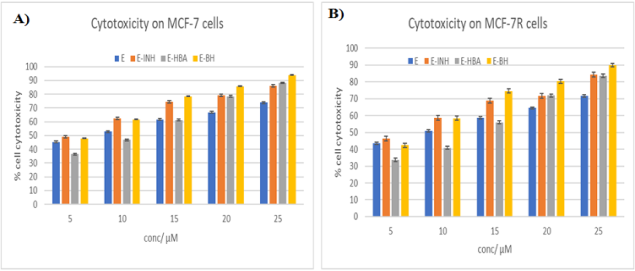
Figure 1. Cytotoxic effects of esculetin derivatives on A) MCF-7 and B) MCF-7 R cells observed by MTT assay for 72 h. [24 and 48 h data are included in the SI].
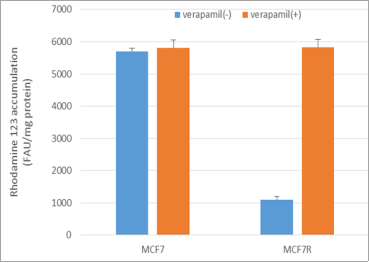
Figure 2. Accumulation of rhodamine 123 in MCF-7 and MCF7R cells. Cells were exposed to 5μM rhodamine 123 for 30 min at 37ºC in the absence or presence of 50μM verapamil and quantified by spectrofluorimetry. Data are expressed as fluorescence arbitrary unit (FAU)/mg protein and are the mean ± SEM of three independent experiments.
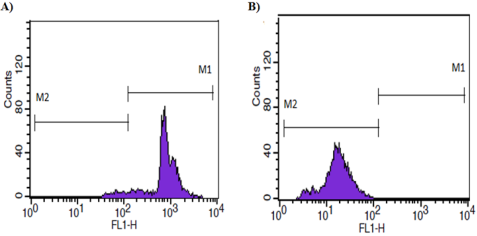
Figure 3. Flow cytometry histogram of the rhodamine 123 accumulation assay; A) sensitive MCF-7 cell line and B) resistant MCF-7 subline. M1 represents the high fluorescence (FL) region, M2 represents the low FL region.
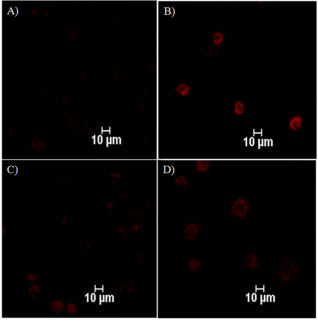
Figure 4. Fluorescence imaging of esculetin derivatives uptake using TRITC filter; A) Esculetin (E), B) E-BH, C) E-HBA and D) E-INH.
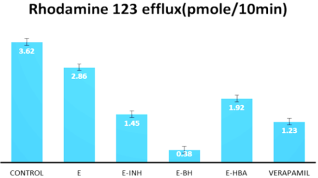
Figure 5. Efflux of rhodamine 123 in MCF7/R cells. Verapamil 5μM was used as a positive control.
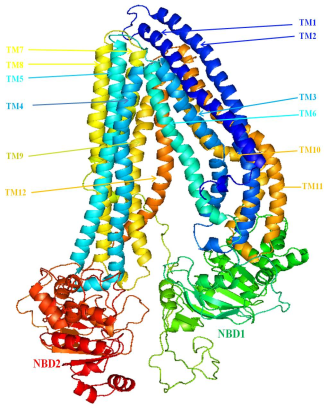
Figure 6. Predicted three-dimensional structure of human P-gp using homology modeling study.
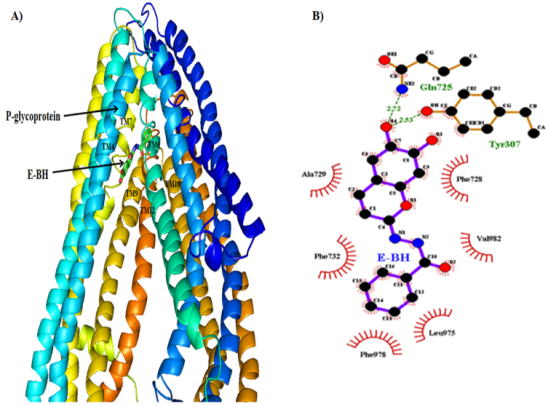
Figure 7. A) Energetically favorable docked complex of P-gp and E-BH, and B) Hydrogen bonding and hydrophobic interactions from favored docked complex of P-gp and E-BH.
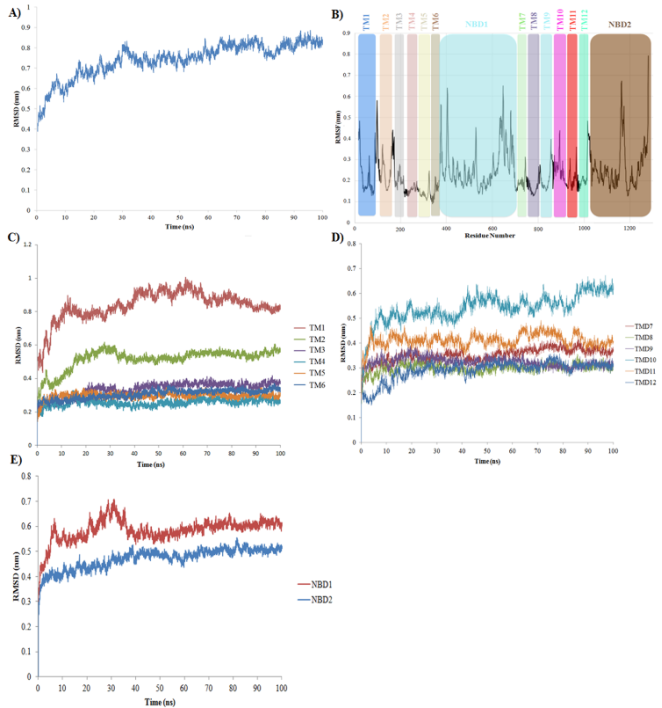
Figure 8. A) RMSD fluctuation of P-gp and E-BH complex during 100 ns of MD simulation study. B) RMSF fluctuations for 1280 residues of human P-gp during 100 ns of simulation, C) RMSD fluctuations in TM helices of TMD1, D) RMSD fluctuations in TM helices of TMD2 and E) RMSD fluctuations in NBD1 and NBD2 of human P-gp.
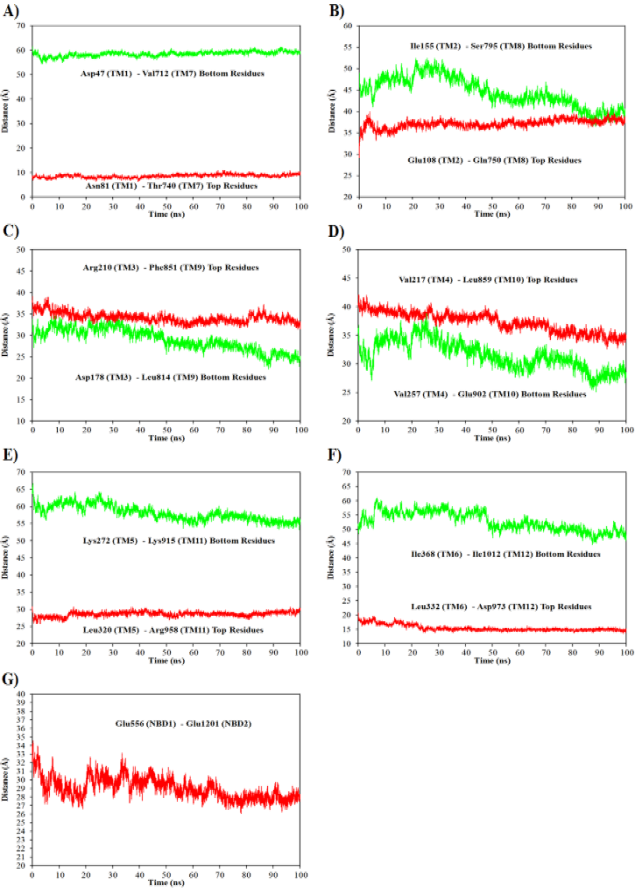
Figure 9. Calculated the distances between the top and bottom residues of adjacent TMs helices facing each other in two TMDs during simulation; A) Distance between the top (red color) and bottom residues (green color) from TM1 and TM7 helices, B) TM2 and TM8, C) TM3 and TM9, D) TM4 and TM10, E) TM5 and TM11, D) TM6 and TM12 and G) Distance between NBD1 and NBD2 domains.
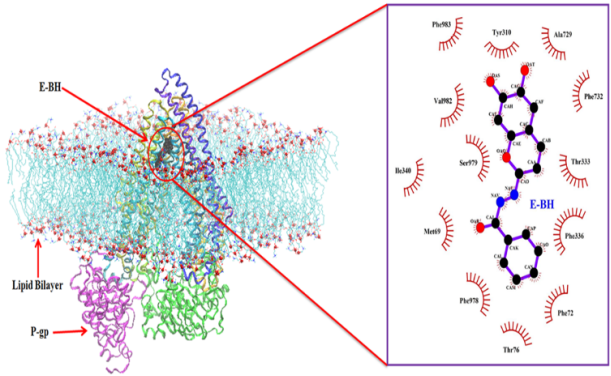
Figure 10. Depicted molecular interactions between E-BH and P-gp after 100 ns of MD simulation study.
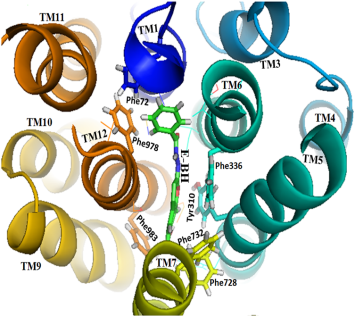
Figure 11. The top view of the human P-gp and E-BH simulated complex showed the interactions from the hydrophobic residues with the E-BH during simulation.
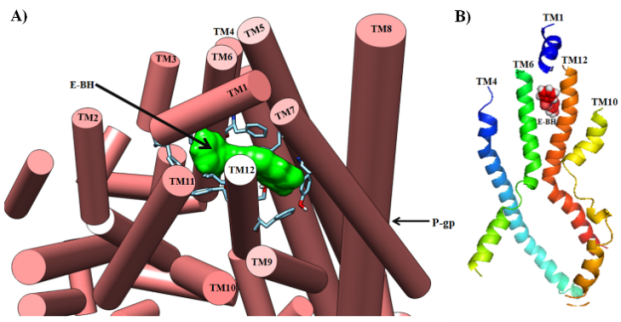
Figure 12. A) Depicted the role of TM1 and TM7 domains in preventing the exit of E-BH from the human P-gp transporter by blocking the exit site. B) The location of E-BH in the interface between transmembrane TM4, TM6, TM10, and TM12 domains and the TM1 domains was blocking the exit site of the P-gp transporter.
Information

