Parkinson disease is a progressive neurodegenerative disorder marked by the abnormal buildup of α-synuclein into toxic fibrils, which lead to neuronal degeneration and motor problems. Among all the identified variants, the type 5A polymorphic structure (8PK4) has been strongly associated with disease progression and represents a promising therapeutic target for the development of safer and more effective drug candidates. In the present study, an integrated computational framework combining with molecular docking and machine-learning-based toxicity prediction was employed to identify potential natural compounds with high therapeutic efficacy and minimal toxic effects. Five bioactive phytochemicals, namely baicalein, rutin, ellagic acid, kaempferol, and ferulic acid, were selected based on their reported neuroprotective potential and screened against the α-synuclein target protein. Molecular docking analysis was performed using the CB-Dock platform to evaluate binding affinity, interaction stability, and residue-level interactions within the active binding pocket. The results demonstrated that all selected compounds exhibited favourable binding interactions with critical amino acid residues, particularly PHE4, LYS6, and GLU35, which are associated with α-synuclein aggregation and stabilization. Among the tested compounds, ellagic acid displayed the strongest binding affinity and the most stable interaction profile, suggesting enhanced inhibitory potential against the target protein. To further assess drug safety, toxicity predictions were performed using the ProTox-II machine-learning platform, evaluating multiple toxicity endpoints, including hepatotoxicity, neurotoxicity, mutagenicity, carcinogenicity, immunotoxicity, and cytochrome P450-mediated interactions. The toxicity assessment revealed that ellagic acid exhibited the lowest predicted toxicity among all screened compounds, while rutin showed a comparatively high LD50 value, indicating reduced acute toxicity and a favourable safety margin. The integration of molecular docking with artificial intelligence-driven toxicity prediction provides a rapid, cost-effective, and reliable strategy for safer drug candidate screening in Parkinson’s disease research. Overall, the study highlights the potential of natural compounds, particularly ellagic acid, as promising therapeutic leads for further experimental validation and future neuroprotective drug development.
| Published in | Computational Biology and Bioinformatics (Volume 14, Issue 1) |
| DOI | 10.11648/j.cbb.20261401.14 |
| Page(s) | 41-53 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Parkinson’s Disease, α-Synuclein, 8PK4 Polymorph, Molecular Docking, Machine Learning, Toxicity Prediction, ProTox-II, Drug Discovery
Target | Baicalein | Rutin | Ferulic acid | Kaempferol | Ellagic acid |
|---|---|---|---|---|---|
LD50 mg/kg | 3919 | 5000 | 1190 | 3919 | 2991 |
Toxicity Class | Class V | Class V | Class IV | Class V | Class IV |
Neurotoxicity | Inactive (0.89) | Inactive (0.89) | Active (0.87) | Inactive (0.89) | Inactive (0.91) |
Respiratory toxicity | Active (0.83) | Active (0.63) | Active (0.98) | Active (0.83) | Active (0.84) |
Mutagenicity | Active (0.51) | Inactive (0.88) | Inactive (0.97) | Inactive (0.52) | Inactive (0.84) |
BBB-barrier | Active (0.53) | Inactive (0.64) | Inactive (1) | Active (0.57) | Inactive (0.90) |
Nutritional toxicity | Inactive (0.53) | Inactive (0.75) | Inactive (0.56) | Active (0.66) | Active (0.60) |
Peroxisome Proliferator Activated Receptor Gamma (PPAR-Gamma) | Active (0.63) | Active (0.54) | Inactive (0.74) | Inactive (0.95) | Active (0.71) |
Nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (nrf2/ARE) | Inactive (0.98) | Inactive (0.98) | Inactive (0.88) | Inactive (0.99) | Inactive (0.99) |
Mitochondrial Membrane Potential (MMP) | Inactive (0.99) | Inactive (0.99) | Inactive (0.70) | Active (1) | Inactive (0.86) |
Phosphoprotein (Tumour Suppressor) p53 | Active (1) | Inactive (0.97) | Inactive (0.96) | Inactive (0.92) | Inactive (0.95) |
GABA receptor (GABAR) | Inactive (0.97) | Inactive (0.90) | Inactive (0.96) | Inactive (0.96) | Inactive (0.66) |
Glutamate N-methyl-D-aspartate receptor (NMDAR) | Inactive (0.96) | Inactive (0.96) | Inactive (0.92) | Inactive (0.92) | Inactive (0.98) |
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor (AMPAR) | Inactive (0.92) | Inactive (0.92) | Inactive (0.97) | Inactive (0.97) | Inactive (1) |
Achetylcholinesterase (AChE) | Inactive (0.69) | Inactive (0.97) | Active (0.69) | Inactive (0.68) | Inactive (0.73) |
Cytochrome CYP2D6 | Inactive (0.85) | Inactive (0.80) | Inactive (0.85) | Active (0.62) | Inactive (0.82) |
Cytochrome CYP3A4 | Inactive (0.79) | Inactive (0.92) | Active (0.79) | Inactive (0.65) | Inactive (0.95) |
| [1] | Hasin, Y.; Seldin, M.; Lusis, A. Multi-Omics Approaches to Disease. Genome Biol. 2017, 18(1), 83. |
| [2] | Kumari, P.; Ghosh, D.; Vanas, A.; Fleischmann, Y.; Wiegand, T.; Jeschke, G.; Riek, R.; Eichmann, C. Structural Insights into α-Synuclein Monomer–Fibril Interactions. Proc. Natl. Acad. Sci. U. S. A. 2021, 118(10), e2012171118. |
| [3] | Singh, N.; Kumari, U. Mult-Omics-Based Drug Discovery Pipelines Incorporating Machine Learning, AI, and BioPython in Parkinson’s Disease: A Case Study of 5A Polymorph of Alpha-Synuclein. 2026, 13(2). |
| [4] | Du, P.; Fan, R.; Zhang, N.; Wu, C.; Zhang, Y. Advances in Integrated Multi-Omics Analysis for Drug-Target Identification. Biomolecules 2024, 14(6), 692. |
| [5] | Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid Fibril Structure of α-Synuclein Determined by Cryo-Electron Microscopy. Cell Res. 2018, 28(9), 897–903. |
| [6] | KUMARI, U.; Pacholee, K. In-Silico Drug Discovery-Based Approach to Treat Impairments in Patients of Alzheimer’s Disease. In JETIR; 2023; Vol. 10. |
| [7] | Zhang, J.; Zheng, M.; Shi, W. Parkinson’s Disease: An Integrative Bioinformatics and Machine Learning Analysis Reveals Tryptophan Metabolism-Associated Gene Biomarkers. BMC Neurol. 2025, 25, 487. |
| [8] | Chaudhary, S.; Kumari, U. NGS, MOLECULAR DOCKING AND NETWORK PHARMACOLOGY REVEAL POTENT INHIBITOR FOR THE TREATMENT OF LUNG CANCER. 2024, 11(9). |
| [9] | Kumari, U. Next-Generation Sequencing (NGS) and Artificial Intelligence for Structural and Functional Analysis of KRAS-G12C in Complex with Novel Inhibitors. 2025, 12(9). |
| [10] | Kumari, U.; Gangurde, D.; Nair, A.; Phirke, N. INTEGRATING COMPUTER AIDED DRUG DESIGN AND NEXT-GENERATION SEQUENCING TO OPTIMIZE TARGETED THERAPIES FOR PI3KΑ H1047R MUTANT IN CANCER. 2025, 12(8). |
| [11] | Kumari, U.; Author), R. P. (First. Structure Analysis and Molecular Docking of Mesothelin-207 Fragment in Human Cancer. In JETIR; 2025; Vol. 12. |
| [12] | Ching, T.; Himmelstein, D. S.; Beaulieu-Jones, B. K.; Kalinin, A. A.; Do, B. T.; Way, G. P.; Ferrero, E.; Agapow, P.-M.; Zietz, M.; Hoffman, M. M.; Xie, W.; Rosen, G. L.; Lengerich, B. J.; Israeli, J.; Lanchantin, J.; Woloszynek, S.; Carpenter, A. E.; Shrikumar, A.; Xu, J.; Cofer, E. M.; Lavender, C. A.; Turaga, S. C.; Alexandari, A. M.; Lu, Z.; Harris, D. J.; DeCaprio, D.; Qi, Y.; Kundaje, A.; Peng, Y.; Wiley, L. K.; Segler, M. H. S.; Boca, S. M.; Swamidass, S. J.; Huang, A.; Gitter, A.; Greene, C. S. Opportunities and Obstacles for Deep Learning in Biology and Medicine. J. R. Soc. Interface 2018, 15(141), 20170387. |
| [13] | Kumari, U.; Pradhan, M.; Mukherjee, S.; Chakrabarti, S. NGS ANALYSIS APPROACH FOR NEURODEGENERATIVE DISEASE WITH BIOPYTHON. In JETIR; 2023; Vol. 10. |
| [14] | Mehrotra, K.; Kumari, U.; Pande, S. Integrative Workflow Of Biopython And Molecuar Docking To Explore Novel Therapeutics Targeting. |
| [15] | Mehra, S.; Gadhe, L.; Bera, R.; Sawner, A. S.; Maji, S. K. Structural and Functional Insights into α-Synuclein Fibril Polymorphism. Biomolecules 2021, 11(10), 1419. |
| [16] | Liu, Y.; Grimm, M.; Dai, W.; Hou, M.; Xiao, Z.-X.; Cao, Y. CB-Dock: A Web Server for Cavity Detection-Guided Protein–Ligand Blind Docking. Acta Pharmacol. Sin. 2020, 41(1), 138–144. |
| [17] | Trott, O.; Olson, A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31(2), 455–461. |
| [18] | Coskuner, O.; Wise-Scira, O. Structures and Free Energy Landscapes of the A53T Mutant-Type α-Synuclein Protein and Impact of A53T Mutation on the Structures of the Wild-Type α-Synuclein Protein with Dynamics. ACS Chem. Neurosci. 2013, 4(7), 1101–1113. |
| [19] | Lamberto, G. R.; Binolfi, A.; Orcellet, M. L.; Bertoncini, C. W.; Zweckstetter, M.; Griesinger, C.; Fernández, C. O. Structural and Mechanistic Basis behind the Inhibitory Interaction of PcTS on α-Synuclein Amyloid Fibril Formation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106(50), 21057–21062. |
| [20] | Sidhu, A.; Segers-Nolten, I.; Raussens, V.; Claessens, M. M. A. E.; Subramaniam, V. Distinct Mechanisms Determine α-Synuclein Fibril Morphology during Growth and Maturation. ACS Chem. Neurosci. 2017, 8(3), 538–547. |
| [21] | Milchberg, M. H.; Warmuth, O. A.; Borcik, C. G.; Dhavale, D. D.; Wright, E. R.; Kotzbauer, P. T.; Rienstra, C. M. Alpha-Synuclein Fibril Structures Cluster into Distinct Classes. bioRxiv 2025, 2025.04.30. 651534. |
| [22] | Sivakumar, P.; Nagashanmugam, K. B.; Priyatharshni, S.; Lavanya, R.; Prabhu, N.; Ponnusamy, S. Review on the Interactions between Dopamine Metabolites and α-Synuclein in Causing Parkinson’s Disease. Neurochem. Int. 2023, 162, 105461. |
| [23] | Hernandez, S. M.; Tikhonova, E. B.; Karamyshev, A. L. Protein-Protein Interactions in Alpha-Synuclein Biogenesis: New Potential Targets in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 72. |
| [24] | Masato, A.; Plotegher, N.; Terrin, F.; Sandre, M.; Faustini, G.; Thor, A.; Adams, S.; Berti, G.; Cogo, S.; De Lazzari, F.; Fontana, C. M.; Martinez, P. A.; Strong, R.; Bandopadhyay, R.; Bisaglia, M.; Bellucci, A.; Greggio, E.; Dalla Valle, L.; Boassa, D.; Bubacco, L. DOPAL Initiates αSynuclein-Dependent Impaired Proteostasis and Degeneration of Neuronal Projections in Parkinson’s Disease. Npj Park. Dis. 2023, 9(1), 42. |
| [25] |
Alpha-synuclein in Parkinson’s disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction | Cell Death & Disease.
https://www.nature.com/articles/s41419-023-05672-9 (accessed 2026-04-25). |
| [26] |
Rostlab/prot_bert Hugging Face.
https://huggingface.co/Rostlab/prot_bert (accessed 2026-04-25). |
| [27] | Shaw, R.; Love, S. D.; McWhite, C. D. Evaluating Pretrained Protein Language Model Embeddings as Proxies for Functional Similarity. J. Mol. Evol. 2025, 93(6), 765–776. |
| [28] | Tule, S.; Foley, G.; Bodén, M. Do Protein Language Models Learn Phylogeny? bioRxiv September 26, 2024, p 2024.09.23. 614642. |
| [29] | Bahar, I.; Lezon, T. R.; Yang, L.-W.; Eyal, E. Global Dynamics of Proteins: Bridging Between Structure and Function. Annu. Rev. Biophys. 2010, 39(1), 23–42. |
| [30] | Banerjee, P.; Eckert, A. O.; Schrey, A. K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46(W1), W257–W263. |
| [31] | Chia, S.; Faidon Brotzakis, Z.; Horne, R. I.; Possenti, A.; Mannini, B.; Cataldi, R.; Nowinska, M.; Staats, R.; Linse, S.; Knowles, T. P. J.; Habchi, J.; Vendruscolo, M. Structure-Based Discovery of Small-Molecule Inhibitors of the Autocatalytic Proliferation of α-Synuclein Aggregates. Mol. Pharm. 2023, 20(1), 183–193. |
| [32] | Ardah, M. T.; Eid, N.; Kitada, T.; Haque, M. E. Ellagic Acid Prevents α-Synuclein Aggregation and Protects SH-SY5Y Cells from Aggregated α-Synuclein-Induced Toxicity via Suppression of Apoptosis and Activation of Autophagy. Int. J. Mol. Sci. 2021, 22(24), 13398. |
| [33] | Kumar, S.; Kumar, R.; Kumari, M.; Kumari, R.; Saha, S.; Bhavesh, N. S.; Maiti, T. K. Ellagic Acid Inhibits α-Synuclein Aggregation at Multiple Stages and Reduces Its Cytotoxicity. ACS Chem. Neurosci. 2021, 12(11), 1919–1930. |
| [34] | Tinku; Choudhary, S. Inhibition of α-Synuclein Fibrillation by Natural Polyphenols: Thermodynamic and Biophysical Aspects. J. Chem. Thermodyn. 2023, 177, 106951. |
APA Style
Singh, N., Kumari, U., Mandra, D., Marouthu, A. (2026). Integrated Machine Learning-based Toxicity Prediction with Molecular Docking for Safer Drug Candidate Screening in Parkinson’s Disease. Computational Biology and Bioinformatics, 14(1), 41-53. https://doi.org/10.11648/j.cbb.20261401.14
ACS Style
Singh, N.; Kumari, U.; Mandra, D.; Marouthu, A. Integrated Machine Learning-based Toxicity Prediction with Molecular Docking for Safer Drug Candidate Screening in Parkinson’s Disease. Comput. Biol. Bioinform. 2026, 14(1), 41-53. doi: 10.11648/j.cbb.20261401.14
@article{10.11648/j.cbb.20261401.14,
author = {Neha Singh and Uma Kumari and Dhanashri Mandra and Aashritha Marouthu},
title = {Integrated Machine Learning-based Toxicity Prediction with Molecular Docking for Safer Drug Candidate Screening in Parkinson’s Disease},
journal = {Computational Biology and Bioinformatics},
volume = {14},
number = {1},
pages = {41-53},
doi = {10.11648/j.cbb.20261401.14},
url = {https://doi.org/10.11648/j.cbb.20261401.14},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cbb.20261401.14},
abstract = {Parkinson disease is a progressive neurodegenerative disorder marked by the abnormal buildup of α-synuclein into toxic fibrils, which lead to neuronal degeneration and motor problems. Among all the identified variants, the type 5A polymorphic structure (8PK4) has been strongly associated with disease progression and represents a promising therapeutic target for the development of safer and more effective drug candidates. In the present study, an integrated computational framework combining with molecular docking and machine-learning-based toxicity prediction was employed to identify potential natural compounds with high therapeutic efficacy and minimal toxic effects. Five bioactive phytochemicals, namely baicalein, rutin, ellagic acid, kaempferol, and ferulic acid, were selected based on their reported neuroprotective potential and screened against the α-synuclein target protein. Molecular docking analysis was performed using the CB-Dock platform to evaluate binding affinity, interaction stability, and residue-level interactions within the active binding pocket. The results demonstrated that all selected compounds exhibited favourable binding interactions with critical amino acid residues, particularly PHE4, LYS6, and GLU35, which are associated with α-synuclein aggregation and stabilization. Among the tested compounds, ellagic acid displayed the strongest binding affinity and the most stable interaction profile, suggesting enhanced inhibitory potential against the target protein. To further assess drug safety, toxicity predictions were performed using the ProTox-II machine-learning platform, evaluating multiple toxicity endpoints, including hepatotoxicity, neurotoxicity, mutagenicity, carcinogenicity, immunotoxicity, and cytochrome P450-mediated interactions. The toxicity assessment revealed that ellagic acid exhibited the lowest predicted toxicity among all screened compounds, while rutin showed a comparatively high LD50 value, indicating reduced acute toxicity and a favourable safety margin. The integration of molecular docking with artificial intelligence-driven toxicity prediction provides a rapid, cost-effective, and reliable strategy for safer drug candidate screening in Parkinson’s disease research. Overall, the study highlights the potential of natural compounds, particularly ellagic acid, as promising therapeutic leads for further experimental validation and future neuroprotective drug development.},
year = {2026}
}
TY - JOUR T1 - Integrated Machine Learning-based Toxicity Prediction with Molecular Docking for Safer Drug Candidate Screening in Parkinson’s Disease AU - Neha Singh AU - Uma Kumari AU - Dhanashri Mandra AU - Aashritha Marouthu Y1 - 2026/06/27 PY - 2026 N1 - https://doi.org/10.11648/j.cbb.20261401.14 DO - 10.11648/j.cbb.20261401.14 T2 - Computational Biology and Bioinformatics JF - Computational Biology and Bioinformatics JO - Computational Biology and Bioinformatics SP - 41 EP - 53 PB - Science Publishing Group SN - 2330-8281 UR - https://doi.org/10.11648/j.cbb.20261401.14 AB - Parkinson disease is a progressive neurodegenerative disorder marked by the abnormal buildup of α-synuclein into toxic fibrils, which lead to neuronal degeneration and motor problems. Among all the identified variants, the type 5A polymorphic structure (8PK4) has been strongly associated with disease progression and represents a promising therapeutic target for the development of safer and more effective drug candidates. In the present study, an integrated computational framework combining with molecular docking and machine-learning-based toxicity prediction was employed to identify potential natural compounds with high therapeutic efficacy and minimal toxic effects. Five bioactive phytochemicals, namely baicalein, rutin, ellagic acid, kaempferol, and ferulic acid, were selected based on their reported neuroprotective potential and screened against the α-synuclein target protein. Molecular docking analysis was performed using the CB-Dock platform to evaluate binding affinity, interaction stability, and residue-level interactions within the active binding pocket. The results demonstrated that all selected compounds exhibited favourable binding interactions with critical amino acid residues, particularly PHE4, LYS6, and GLU35, which are associated with α-synuclein aggregation and stabilization. Among the tested compounds, ellagic acid displayed the strongest binding affinity and the most stable interaction profile, suggesting enhanced inhibitory potential against the target protein. To further assess drug safety, toxicity predictions were performed using the ProTox-II machine-learning platform, evaluating multiple toxicity endpoints, including hepatotoxicity, neurotoxicity, mutagenicity, carcinogenicity, immunotoxicity, and cytochrome P450-mediated interactions. The toxicity assessment revealed that ellagic acid exhibited the lowest predicted toxicity among all screened compounds, while rutin showed a comparatively high LD50 value, indicating reduced acute toxicity and a favourable safety margin. The integration of molecular docking with artificial intelligence-driven toxicity prediction provides a rapid, cost-effective, and reliable strategy for safer drug candidate screening in Parkinson’s disease research. Overall, the study highlights the potential of natural compounds, particularly ellagic acid, as promising therapeutic leads for further experimental validation and future neuroprotective drug development. VL - 14 IS - 1 ER -
Bioinformatics Project and Research Institute, Noida, India
Bioinformatics Project and Research Institute, Noida, India
Bioinformatics Project and Research Institute, Noida, India
Bioinformatics Project and Research Institute, Noida, India
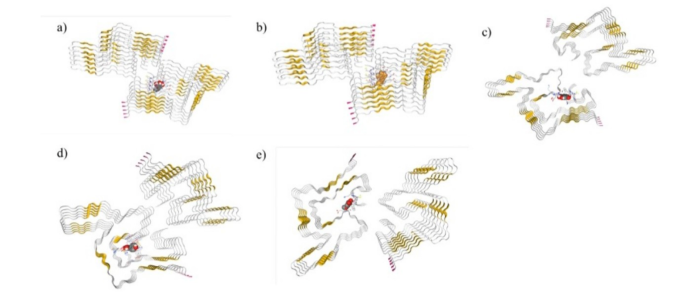
Figure 1. Molecular docking interactions of selected bioactive compounds with α-synuclein polymorph 8PK4, a) Baicalein, b) Rutin, c) Ellagic acid, d) Kaempferol e) Ferulic acid docked within the predicted binding cavities of the protein. The dominance of α-helices suggests structural rigidity and functional specificity. The interaction pattern (hydrogen bonds + hydrophobic contacts) indicates: Strong binding affinity, structure is dominated by α-helices (yellow regions) connected by loops (grey). The central region contains a ligand (grey spherical model).
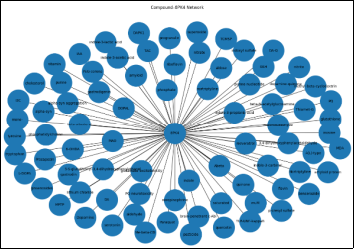
Figure 2. Compound-protein interaction network of 8PK4 showing interactions with diverse molecules, including neurotransmitters, oxidative stress markers, and bioactive compounds, highlighting its central role in Parkinson’s disease-associated pathways.
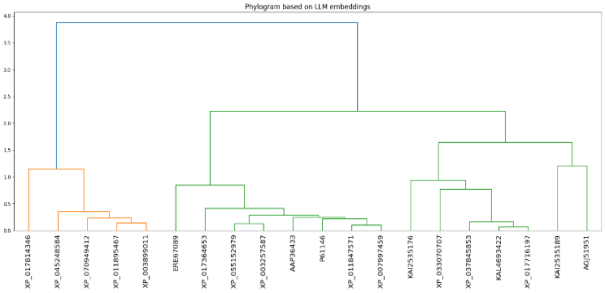
Figure 3. Phylogenetic tree (dendrogram) clustering of protein sequences associated with alpha synuclein polymorphic variance (8PK4) was conducted using an integrated transformer-derived embedding method (i.e., ProtBERT and ESM). Clustering was completed using Ward's minimum variance clustering method, with branch distances indicating the degree of difference between protein sequences. Closely spaced protein sequences in the same cluster have similar structure and evolution. Distantly spaced protein sequences are likely to be different due to protein structural features. [31, 32].
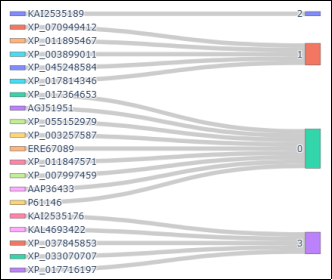
Figure 4. 8PK4 protein clustering from an automated or machine learning-based embedding analysis (with examples of homologous/substituted sequences being compared) is shown in the Sankey diagram to the left. Protein sequences on the left are individual proteins (nodes) and those on the right are clusters of like proteins (nodes). The Sankey diagram depicts the flow of these protein sequences to the clusters they are associated with, as well as the major clusters of similar protein sequences as interpreted by the machine-learning or substituted sequence analysis.
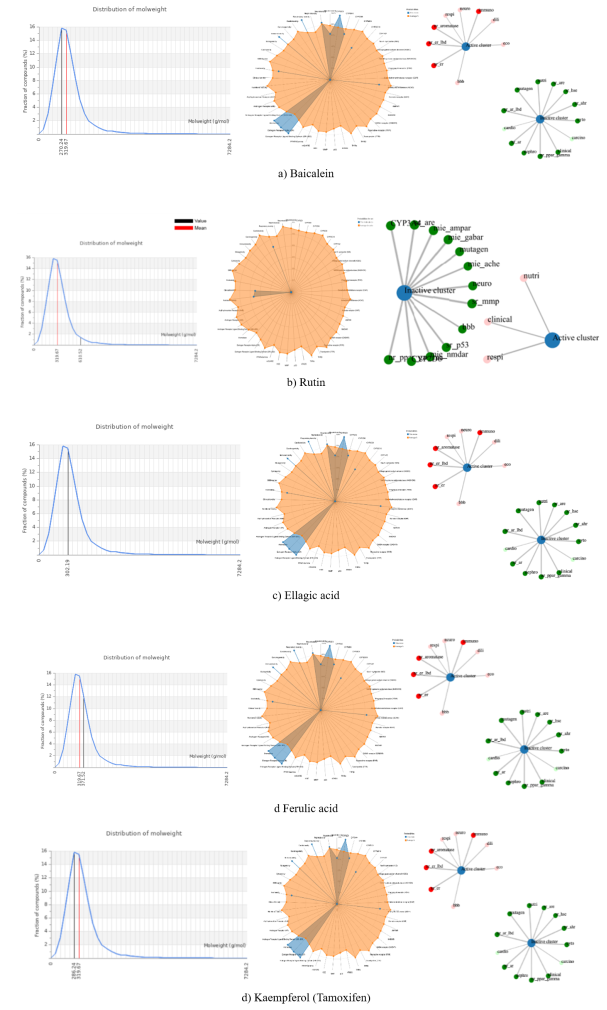
Figure 5.
Comprehensive physicochemical and toxicity profiling of the studied compounds. The left panel shows the molecular weight distribution histograms, indicating a narrow distribution with mean values highlighted (red line). The middle panel presents radar plots summarising key drug-likeness and ADMET properties, demonstrating that most compounds fall within acceptable ranges. The right panel illustrates toxicity prediction networks, where compounds are clustered based on biological activity, highlighting potential toxicological endpoints (e.g., mutagenicity, carcinogenicity, and irritancy).Information

