Chronic myeloid leukaemia (CML) is a clonal myeloproliferative cancer caused by the constant activity of the BCR–ABL 1 fusion protein' s tyrosine kinase, resulting from the Philadelphia chromosome translocation, which leads to abnormal cell growth, survival, and disease progression. While tyrosine kinase inhibitors (TKIs) have greatly improved patient outcomes, issues like drug resistance and persistent leukemic stem cells highlight the need for alternative therapies. This study used a structure- guided CRISPR- Cas 9 genome editing approach to identify highly specific single- guide RNAs (sgRNAs) that can disrupt the human ABL 1 gene, a key part of the BCR–ABL 1 fusion. The high- resolution crystal structure of the ABL 1 kinase domain (PDB ID: 8 I 7 S) helped identify essential functional regions, including catalytic and ATP- binding sites, for precise CRISPR targeting. Computational design and filtering of sgrnas were performed using E- CRISP and CHOPCHOP, focusing on criteria like PAM site accessibility, targeting conserved kinase regions in exons, GC content, predicted efficiency, and low off- target risk. In silico analyses, including specificity scores, mismatch profiles, and sequence alignment across ABL 1 transcript variants, ensured high selectivity and broad coverage. Genomic visualization confirmed accurate targeting within exons encoding vital kinase functions. Protein–protein interaction analysis via STRING showed strong links between ABL 1 and key oncogenic regulators such as BCR, STAT 5, and MAPK pathway components. KEGG pathway analysis further indicated ABL 1' s involvement in chronic myeloid leukaemia, PI 3 K–AKT, MAPK signaling, and other cancer- related pathways, emphasising its importance in CML development. This combined computational approach demonstrates that structure- guided CRISPR- Cas 9 targeting of ABL 1 can effectively disrupt BCR–ABL 1 driven cancer signals. The results provide a strong theoretical basis for future experimental validation and genome editing therapies aimed at overcoming TKI resistance and achieving long- lasting molecular remission in CML.
| Published in | Computational Biology and Bioinformatics (Volume 14, Issue 1) |
| DOI | 10.11648/j.cbb.20261401.12 |
| Page(s) | 13-25 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
CRISPR-Cas9, ABL1, BCR-ABL, Chronic Myeloid Leukaemia, sgRNA Design, E-CRISP, CHOPCHOP, Off-target Analysis
ABL1 | Abelson Murine leukemia Viral Oncogene Homolog 1 |
BCR | Breakpoint Cluster Region |
BCR–ABL1 | Breakpoint Cluster Region–Abelson 1 Fusion Gene |
BLAST | Basic Local Alignment Search Tool |
Cas9 | CRISPR-Associated Protein 9 |
CML | Chronic Myeloid Leukemia |
CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
DNA | Deoxyribonucleic Acid |
dsDNA | Double-Stranded Deoxyribonucleic Acid |
ERK | Extracellular Signal-Regulated Kinase |
GC | Guanine–Cytosine |
InterProScan | Integrated Protein Signature Recognition Tool |
JAK–STAT | Janus Kinase–Signal Transducer and Activator of Transcription |
MAPK | Mitogen-Activated Protein Kinase |
PAM | Protospacer Adjacent Motif |
PI3K–AKT | Phosphoinositide 3-kinase–Protein Kinase B Signaling Pathway |
PPI | Protein-Protein Interaction |
sgRNA | Single-Guide RNA |
STRING | Search Tool for the Retrieval of Interacting Genes/Proteins |
TCGA | The Cancer Genome Atlas |
TKI | Tyrosine Kinase Inhibitor |
| [1] | Holyoake, T. L., & Vetrie, D. (2017). The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood, 129(12), 1595–1606. |
| [2] | Hochhaus, A., Larson, R. A., Guilhot, F., et al. (2023). Long-term outcomes of tyrosine kinase inhibitor therapy in chronic myeloid leukemia. Leukemia, 37(1), 1–13. |
| [3] | Skorski, T. (2018). BCR-ABL1 kinase: Hunting an elusive target with new weapons. Blood, 132(22), 2369–2374. |
| [4] | Van Etten, R. A. (2020). Mechanisms of transformation by the BCR-ABL oncogene. Hematology/Oncology Clinics of North America, 34(2), 271–285. |
| [5] | Greuber, E. K., Smith-Pearson, P., Wang, J., & Pendergast, A. M. (2013). Role of ABL family kinases in cancer: From leukemia to solid tumors. Nature Reviews Cancer, 13(8), 559–571. |
| [6] | Hantschel, O. (2012). Structure, regulation, and inhibition of BCR-ABL. Journal of Hematology & Oncology, 5, 10. |
| [7] | Jabbour, E., & Kantarjian, H. (2020). Chronic myeloid leukemia: 2020 update on diagnosis, therapy, and monitoring. American Journal of Hematology, 95(6), 691–709 |
| [8] | Awad, M. M., Liu, S., Rybkin, I. I., et al. (2024). CRISPR functional genomics identifies lineage-specific dependencies in chronic myeloid leukemia. Leukemia, 38(2), 312–325. |
| [9] | Tsai, S. Q., & Joung, J. K. (2016). Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics, 17(5), 300–312. |
| [10] | Anzalone, A. V., Koblan, L. W., & Liu, D. R. (2019). Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nature Biotechnology, 38(7), 824–844. |
| [11] | Somuncu, E., Kirmizis, A., & Gozuacik, D. (2025). CRISPR-based functional genomics in hematological malignancies. Frontiers in Oncology, 15, 1298457. |
| [12] | Doench, J. G., Fusi, N., Sullender, M., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology, 34(2), 184–191. |
| [13] | Heigwer, F., Kerr, G., & Boutros, M. (2014). E-CRISP: Fast CRISPR target site identification. Nature Methods, 11(2), 122–123. |
| [14] | Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., & Valen, E. (2019). CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Research, 47(W1), W171–W174. |
| [15] | Ji, J., Zhang, Y., Redmond, D., et al. (2025). Precision CRISPR design strategies for therapeutic genome editing. Trends in Biotechnology, 43(2), 230–245. |
| [16] | Szklarczyk, D., Gable, A. L., Nastou, K. C., et al. (2021). The STRING database in 2021: Customizable protein–protein networks and functional characterization. Nucleic Acids Research, 49(D1), D605–D612. |
| [17] | Shah, N. P., Nicoll, J. M., Nagar, B., Gorre, M. E., Paquette, R. L., Kuriyan, J., & Sawyers, C. L. (2002). Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell, 2(2), 117–125. |
| [18] | Bandbe, T., Johri, V., Kumari, U. (2025). Structure-guided Genome-wide Association Analysis of ALK Variants with GWAS Data Using R. Computational Biology and Bioinformatics, 13(2), 72-85. |
| [19] | Uma Kumari, Gunika Nagpal, Gaurav Verma"CRISPR-Cas9: Revolutionizing Genome Editing and its Applications", International Journal of Emerging Technologies and Innovative Research, Vol 12, Issue 1, page no. h226-h235, January 2025 |
| [20] | Uma Kumari, Narotham BP "Integrative CRISPR-Cas9 and machine learning Approaches for Target Discovery and Therapeutic Development in Malignant Brain Tumor", International Journal of Emerging Technologies and Innovative Research Vol. 12, Issue 12, page no. ppc749-c759, December-2025, |
| [21] | Kaur, J., Konar, A., Halder, S., Chakraborty, S., & Ghosh, P. (2024). CRISPR-Cas9 to treat chronic myeloid leukemia: A review. International Journal of Clinical Biology and Biochemistry, 6(1), 31–36. |
| [22] | Uma Kumari, Shivangi Koundal, and Sumita Katal "Integrated structural and network-based characterization of ABL1 as a central hub in leukemia, using protein interaction and motif analysis with Biopython", International Journal of Emerging Technologies and Innovative Research, Vol. 12, Issue 12, pages g355-g366, December 2025, January 2026. |
APA Style
Katal, S., Koundal, S., Kumari, U. (2026). Structure-guided CRISPR CAS9 Targeting of ABL1 for Functional Disruption of BCR-ABL1 Fusion in Chronic Myeloid Leukaemia. Computational Biology and Bioinformatics, 14(1), 13-25. https://doi.org/10.11648/j.cbb.20261401.12
ACS Style
Katal, S.; Koundal, S.; Kumari, U. Structure-guided CRISPR CAS9 Targeting of ABL1 for Functional Disruption of BCR-ABL1 Fusion in Chronic Myeloid Leukaemia. Comput. Biol. Bioinform. 2026, 14(1), 13-25. doi: 10.11648/j.cbb.20261401.12
@article{10.11648/j.cbb.20261401.12,
author = {Sumita Katal and Shivangi Koundal and Uma Kumari},
title = {Structure-guided CRISPR CAS9 Targeting of ABL1 for Functional Disruption of BCR-ABL1 Fusion in Chronic Myeloid Leukaemia},
journal = {Computational Biology and Bioinformatics},
volume = {14},
number = {1},
pages = {13-25},
doi = {10.11648/j.cbb.20261401.12},
url = {https://doi.org/10.11648/j.cbb.20261401.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cbb.20261401.12},
abstract = {Chronic myeloid leukaemia (CML) is a clonal myeloproliferative cancer caused by the constant activity of the BCR–ABL 1 fusion protein' s tyrosine kinase, resulting from the Philadelphia chromosome translocation, which leads to abnormal cell growth, survival, and disease progression. While tyrosine kinase inhibitors (TKIs) have greatly improved patient outcomes, issues like drug resistance and persistent leukemic stem cells highlight the need for alternative therapies. This study used a structure- guided CRISPR- Cas 9 genome editing approach to identify highly specific single- guide RNAs (sgRNAs) that can disrupt the human ABL 1 gene, a key part of the BCR–ABL 1 fusion. The high- resolution crystal structure of the ABL 1 kinase domain (PDB ID: 8 I 7 S) helped identify essential functional regions, including catalytic and ATP- binding sites, for precise CRISPR targeting. Computational design and filtering of sgrnas were performed using E- CRISP and CHOPCHOP, focusing on criteria like PAM site accessibility, targeting conserved kinase regions in exons, GC content, predicted efficiency, and low off- target risk. In silico analyses, including specificity scores, mismatch profiles, and sequence alignment across ABL 1 transcript variants, ensured high selectivity and broad coverage. Genomic visualization confirmed accurate targeting within exons encoding vital kinase functions. Protein–protein interaction analysis via STRING showed strong links between ABL 1 and key oncogenic regulators such as BCR, STAT 5, and MAPK pathway components. KEGG pathway analysis further indicated ABL 1' s involvement in chronic myeloid leukaemia, PI 3 K–AKT, MAPK signaling, and other cancer- related pathways, emphasising its importance in CML development. This combined computational approach demonstrates that structure- guided CRISPR- Cas 9 targeting of ABL 1 can effectively disrupt BCR–ABL 1 driven cancer signals. The results provide a strong theoretical basis for future experimental validation and genome editing therapies aimed at overcoming TKI resistance and achieving long- lasting molecular remission in CML.},
year = {2026}
}
TY - JOUR T1 - Structure-guided CRISPR CAS9 Targeting of ABL1 for Functional Disruption of BCR-ABL1 Fusion in Chronic Myeloid Leukaemia AU - Sumita Katal AU - Shivangi Koundal AU - Uma Kumari Y1 - 2026/02/04 PY - 2026 N1 - https://doi.org/10.11648/j.cbb.20261401.12 DO - 10.11648/j.cbb.20261401.12 T2 - Computational Biology and Bioinformatics JF - Computational Biology and Bioinformatics JO - Computational Biology and Bioinformatics SP - 13 EP - 25 PB - Science Publishing Group SN - 2330-8281 UR - https://doi.org/10.11648/j.cbb.20261401.12 AB - Chronic myeloid leukaemia (CML) is a clonal myeloproliferative cancer caused by the constant activity of the BCR–ABL 1 fusion protein' s tyrosine kinase, resulting from the Philadelphia chromosome translocation, which leads to abnormal cell growth, survival, and disease progression. While tyrosine kinase inhibitors (TKIs) have greatly improved patient outcomes, issues like drug resistance and persistent leukemic stem cells highlight the need for alternative therapies. This study used a structure- guided CRISPR- Cas 9 genome editing approach to identify highly specific single- guide RNAs (sgRNAs) that can disrupt the human ABL 1 gene, a key part of the BCR–ABL 1 fusion. The high- resolution crystal structure of the ABL 1 kinase domain (PDB ID: 8 I 7 S) helped identify essential functional regions, including catalytic and ATP- binding sites, for precise CRISPR targeting. Computational design and filtering of sgrnas were performed using E- CRISP and CHOPCHOP, focusing on criteria like PAM site accessibility, targeting conserved kinase regions in exons, GC content, predicted efficiency, and low off- target risk. In silico analyses, including specificity scores, mismatch profiles, and sequence alignment across ABL 1 transcript variants, ensured high selectivity and broad coverage. Genomic visualization confirmed accurate targeting within exons encoding vital kinase functions. Protein–protein interaction analysis via STRING showed strong links between ABL 1 and key oncogenic regulators such as BCR, STAT 5, and MAPK pathway components. KEGG pathway analysis further indicated ABL 1' s involvement in chronic myeloid leukaemia, PI 3 K–AKT, MAPK signaling, and other cancer- related pathways, emphasising its importance in CML development. This combined computational approach demonstrates that structure- guided CRISPR- Cas 9 targeting of ABL 1 can effectively disrupt BCR–ABL 1 driven cancer signals. The results provide a strong theoretical basis for future experimental validation and genome editing therapies aimed at overcoming TKI resistance and achieving long- lasting molecular remission in CML. VL - 14 IS - 1 ER -
Department of Bioinformatics, Bioinformatics Project and Research Institute, Noida, India
Department of Bioinformatics, Bioinformatics Project and Research Institute, Noida, India
Department of Bioinformatics, Bioinformatics Project and Research Institute, Noida, India
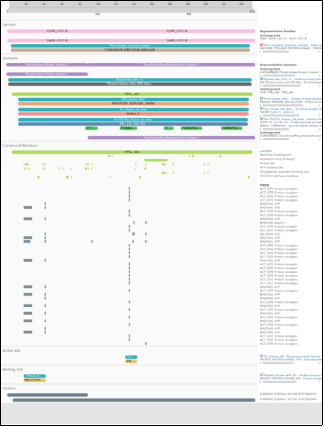
Figure 1. InterProScan-Based Family Classification and Domain Architecture of ABL1.
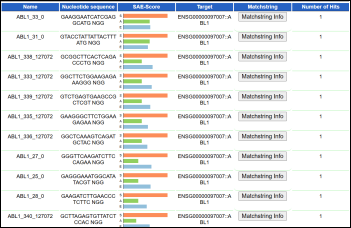
Figure 2. The Output of CRISPR guide RNA (gRNA) target prediction.

Figure 3. Comparative Off-Target and On-Target Efficiency Analysis of ABL1 CRISPR gRNAs.
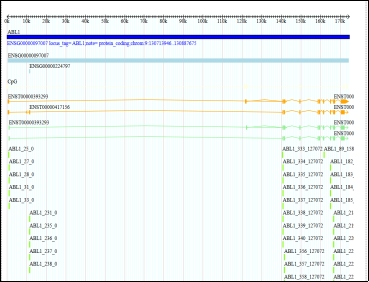
Figure 4. Display of genomic browser, illustrating the ABL1 gene structure.
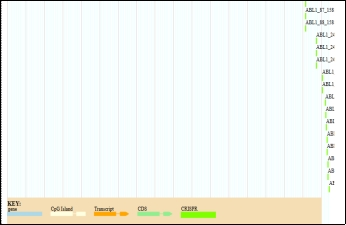
Figure 5. Genomic Organization of the ABL1 Locus Showing Transcript Variants, CpG Islands, and CRISPR Sites.
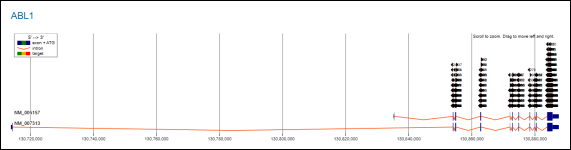
Figure 6. CHOPCHOP-Based Genomic Mapping of CRISPR Targetable Regions within the ABL1 Gene.
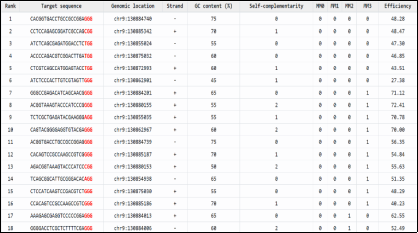
Figure 7. CHOPCHOP Ranking and Characterization of High-Confidence CRISPR–Cas9 sgRNA Targets on Chromosome 9.
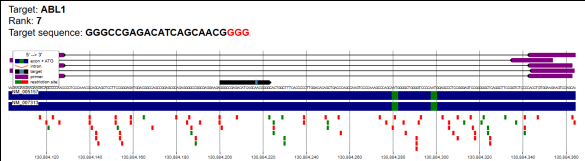
Figure 8. Genome-Wide Off-Target Profiling of the Selected CRISPR–Cas9 sgRNA Targeting ABL1.
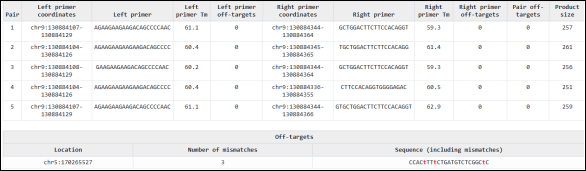
Figure 9. In Silico Design and Specificity Analysis of PCR Primer Pairs Targeting the ABL1 Genomic Region.
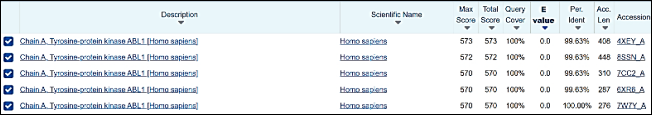
Figure 10. Sequence Similarity search in BLAST.

Figure 11. Protein–Protein Interaction Network of ABL1 and Associated Downstream Signaling Proteins.
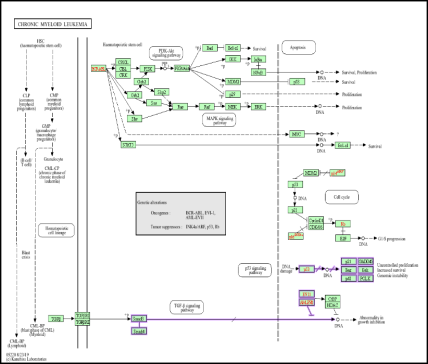
Figure 12. Mapping of BCR-ABL/ABL1-Associated Signaling Cascades in Chronic Myeloid Leukemia.
Information

